

单原子催化剂(Single-Atom Catalysts, SACs)是一类新型的高效催化剂,其结合了均相和非均相催化剂的优点。单原子催化剂具有独特的物理化学性质,如高活性、高选择性和高稳定性,因而在催化领域引起了广泛关注。
本文就几种基于X射线的单原子表征手段展开了讨论:
XAS技术通过测量样品对特定能量X射线的吸收来获取原子结构、电荷状态和元素间相互作用的信息。
该技术一般可以分为硬XAS(hard-XAS)和软XAS(sXAS)两种,其中硬XAS主要用于探测金属中心的电子结构,而软XAS则对轻元素(如C、N、O)的电子态和配位环境更为敏感。
同步辐射技术在研究单原子催化中发挥了关键作用,通过识别催化剂结构和理解反应机制来推动单原子催化的发展。
hard-XAS:
硬XAS利用高能X射线(通常 > 5 keV,如Cu K-edge: 8.98 keV)激发原子内层电子(如1s电子)至未占据的电子态或连续态,通过分析吸收边附近光谱特征,可以获得电子结构、配位环境以及几何结构等相关信息。
硬XAS通过XANES和EXAFS提供了SACs的“电子结构-配位环境”指纹,是验证单原子特性(如孤立性、配位数)和揭示构-效关系的不可替代工具。结合原位手段,可进一步揭示催化反应的真实活性位点。
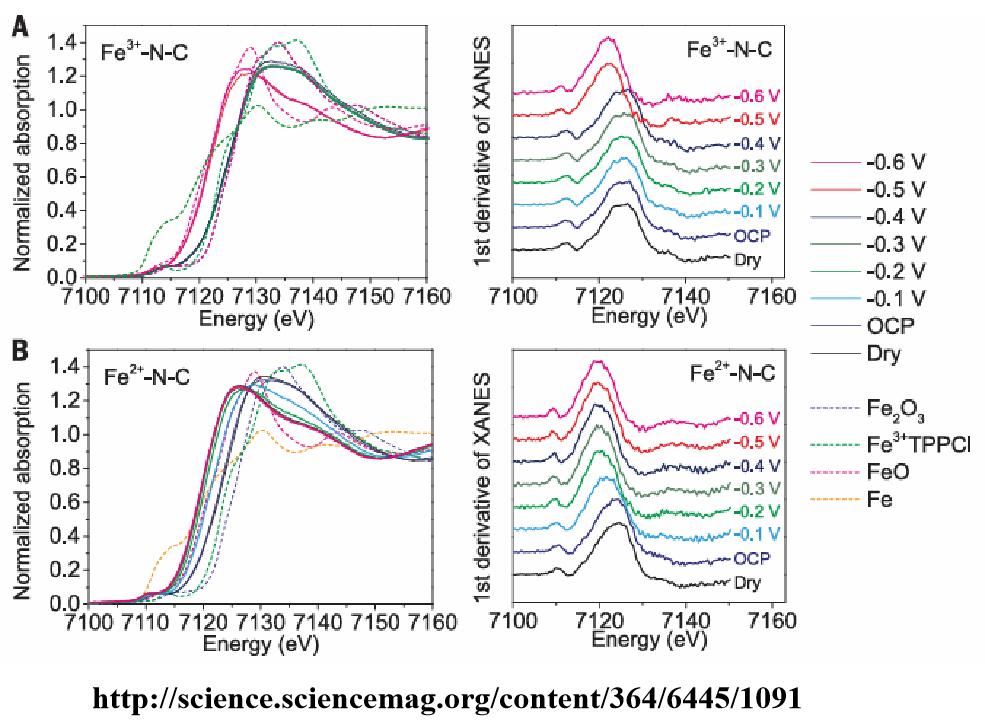

该图为Fe3+-N-C和Fe2+-N-C催化剂在不同电位下的原位X射线吸收光谱,包括X射线吸收近边结构(XANES)及其一阶导数。由图可知,Fe3+-N-C在-0.4 V 时能够保持Fe3+的氧化态,这与催化剂的高活性密切相关。
当电位负移至-0.5 V 时,Fe3+被还原为Fe2+,催化剂的活性降低,这表明Fe3+位点在CO2还原过程中更为活跃。Fe2+-N-C在较低的电位下就开始还原,且在反应条件下主要以Fe2+存在,这解释了其较低的活性和稳定性。
Fe2+-N-C的活性位点在高过电位下受到CO脱附的限制,而Fe3+-N-C则没有这种限制,因此能够在更高的电流密度下运行。
通过原位XAS技术揭示了Fe3+-N-C和Fe2+-N-C在电化学CO2还原过程中的动态变化,明确了Fe3+位点的稳定性和活性优势,为理解催化剂的高性能提供了关键的结构和电子态信息。
sXAS:
软XAS利用低能X射线(200–2000 eV)激发原子中价电子或浅层核心电子(如2p、3d轨道),通过测量X射线吸收系数获得未占据电子态信息。
sXAS作为一种表面敏感技术,能够探测SACs中金属中心的电子态以及配体环境中的轻元素(如C、N、O等)。sXAS能够通过K边吸收光谱来探测这些轻元素的结构变化,尤其是在反应过程中吸附/脱附反应中间体时的变化。
软XAS通过L-edge和K-edge分析,为SACs提供了电子结构-配位化学的原子级洞察,尤其在轻元素配体(如C/N/O)与3d金属相互作用的研究中不可替代。结合原位技术,可动态追踪催化反应中单原子位点的真实状态,从而深入理解催化反应机制。
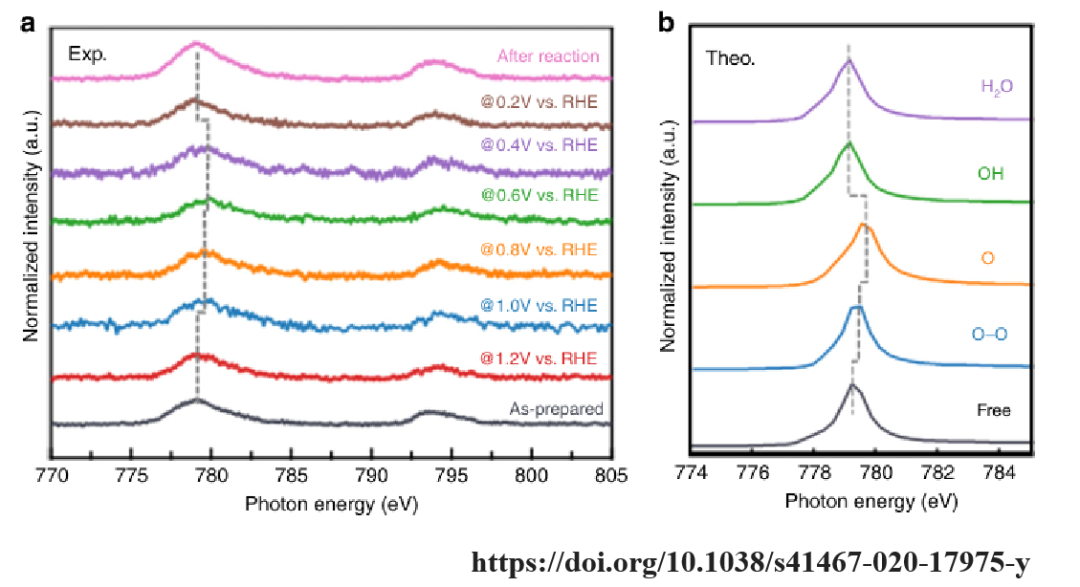
该图为不同电位下,Co L3,2边X射线吸收近边结构(XANES)光谱以及理论计算结果。在1.2 V时,Co L3,2边的特征峰与原始样品相似,表明此时Co中心处于预催化状态,氧分子尚未发生化学吸附。
当电位降低到1.0 V至0.4 V时,Co L3,2边的特征峰向高光子能量方向移动(约1 eV),这表明在这些电位下发生了化学反应,Co中心的氧化态发生了变化。在0.2 V时,特征峰回到原始位置,表明最终产物已经从Co中心脱附。
计算结果表明,氧分子吸附在Co中心时,Co L3,2边的特征峰向高能量方向移动,与实验观察一致。说明氧分子的吸附导致Co中心的氧化态变化,从而促进了氧分子的还原。
通过Co L3,2边XANES光谱和理论计算,揭示了Co-Nx/C催化剂在ORR过程中Co中心的氧化态变化和氧物种的吸附解离机制,为设计高性能非贵金属ORR催化剂提供了重要的理论依据。
原位实验能够实时观察SACs在反应条件下的动态变化,有助于深入理解反应机制和结构-性能关系。尽管如此,原位实验的设计和实施仍面临诸多挑战,如反应环境的模拟和信号干扰问题。
XES作为一种强大的光谱技术,在单原子催化剂(SACs)的研究中具有重要的应用价值。
它不仅能够识别SACs中的配体原子,还能提供关于氧化态、自旋态和电子轨道的信息。此外,XES还可以用于探索配位数和中间体吸附模式,这对于理解SACs的催化活性和反应机制至关重要。
尽管XES在实际应用中面临一些挑战,如低信噪比和复杂的配位环境,但通过结合其他表征技术和理论计算,可以有效克服这些困难,从而为SACs的研究提供更全面和准确的结构信息。
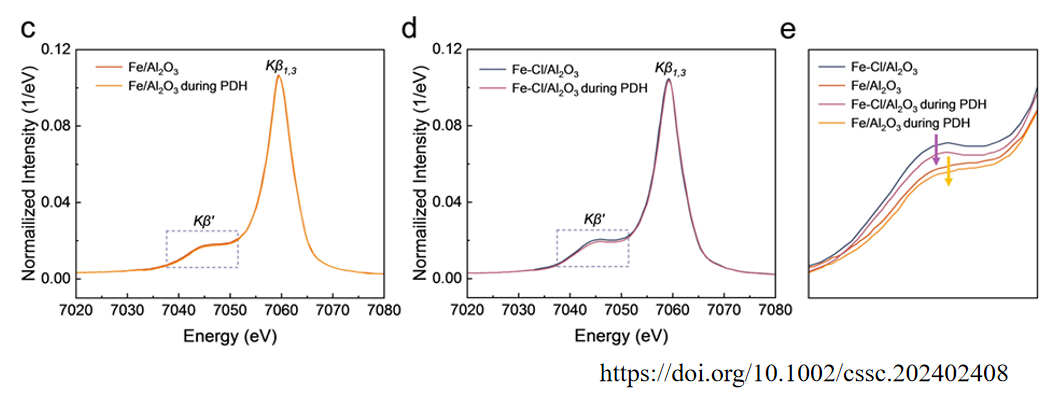
该论文研发了一种新型的单原子铁催化剂(Fe-Cl/Al2O3),通过在Al2O3的Al3+空位中锚定孤立的Fe-Cl位点,显著提高了丙烷脱氢(PDH)反应中的稳定性和活性。
上图展示了Fe/Al2O3和Fe-Cl/Al2O3催化剂在PDH反应中的原位XES光谱。
在Fe-Cl/Al2O3催化剂中,Fe 3d轨道的单电子占据数减少得更为显著,表明Fe-Cl位点在反应过程中具有更强的电子转移能力。
这种电子转移能力的增强有助于降低C3H8转化为*C3H7的能量障碍,从而提高催化活性。图e是Kβ峰的局部放大图,进一步展示了Fe-Cl/Al2O3催化剂在电子转移方面的优势。
这些结果表明,Fe-Cl位点在高自旋态下单原子Fe的3d轨道能够更有效地将电子转移到C3H8的分子轨道中,从而促进C-H键的活化。
随着SACs的不断发展,研究者们发现仅靠硬XAS、sXAS和XES提供的信息仍然不足以深入研究其内在特性。
为了超越稳态或单一技术测量的限制,应用多种光谱技术的组合是一个有前景的方法。例如,共振非弹性X射线散射(RIXS/RXES)提供了比传统XAS和XES更高维度的数据分析,能够更全面地研究电子结构和配位环境。
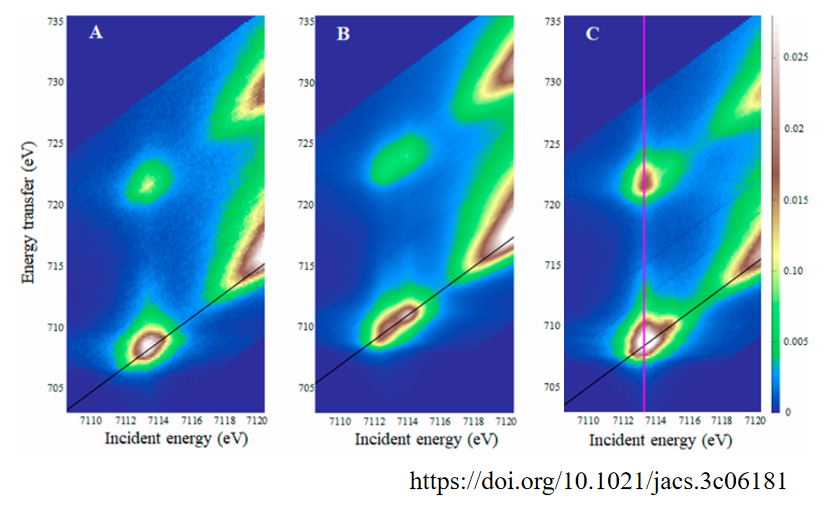
该图展示了[Fe(III)(OH)(H3buea)]−、[Fe(III)O(H3buea)]2−和[Fe(IV)O(H3buea)]−三种复合物的1s2p RIXS平面图。
RIXS图中,横坐标为入射光子能量,纵坐标为能量转移,即入射光子和发射光子之间的能量差。通过CEE(RIXS平面的对角线切割,如图中的黑线所示)穿过RIXS平面可以得到类似铁K边的光谱,其能量分辨率高于标准的K边XAS实验。
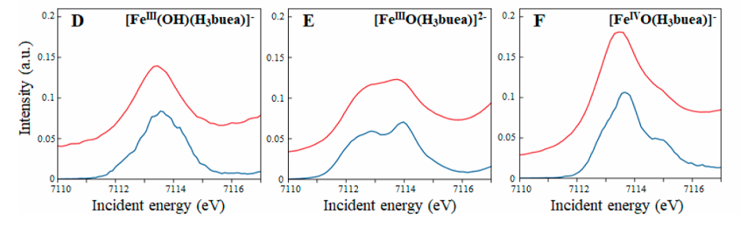
D-F图为三种物质对应的CEE切割(蓝色)和Fe K边XAS光谱(红色),由图可知,CEE切割得到的光谱能量分辨率明显高于标准的K边XAS实验。
图D在7113.5 eV处有一个强烈的预边特征,表明1s电子跃迁到3deσ轨道。该复合物具有D3h对称性,导致Fe 4px和4py轨道与3dxy和3dx²−y²轨道混合,增加了预边强度。
图E在7112.9 eV和7113.9 eV处有两个明显的预边特征,表明1s电子跃迁到3deσ和3dz²轨道。该复合物具有C3v对称性,导致Fe 4pz轨道与3dz²轨道混合,增加了1s→3dz²跃迁的电偶极子强度。
图F在7112.9 eV、7113.6 eV和7114.7 eV处有三个明显的预边特征,表明1s电子跃迁到3deσ、3dz²α和3dz²β轨道。
由于Fe(IV)=O中间体的高自旋态(S = 2),3dz²轨道的α和β自旋态分裂,导致了两个不同的z²跃迁。
该实验通过1s2p RIXS和Fe K边XAS技术详细揭示了Fe(IV)=O中间体的电子结构和几何结构,特别是4p轨道与3d轨道的混合以及自旋态的影响。这些发现为理解Fe(IV)=O中间体的反应性提供了重要的实验依据。
基于X射线的原位表征技术为单原子催化剂(SACs)的研究提供了强大的工具。通过X射线吸收光谱(XAS)、X射线发射光谱(XES)以及共振非弹性X射线散射(RIXS/RXES)等方法,研究人员能够在反应条件下实时监测催化剂的结构和电子态变化。
这些技术不仅帮助揭示了SACs的活性位点和反应机制,还为优化催化剂性能和设计新型高效催化剂提供了重要依据。
未来,结合机器学习和多技术集成的原位表征方法将进一步推动单原子催化领域的研究进展。