

说明:本文华算科技旨在系统阐述蒙特卡洛(Monte Carlo)模拟在材料与化学领域的理论基础、核心算法、适用范围及具体实践方法。
通过对定义、应用案例和操作步骤的详细介绍,辅以图示说明,期望为相关领域的科研人员和学生提供一个清晰、全面的入门指南。
什么是蒙特卡洛模拟
蒙特卡洛(Monte Carlo, MC)模拟并非指某种单一的、确定的算法,而是一类依赖于重复随机抽样和统计分析来解决问题的计算方法的总称。其核心思想是将一个难以直接解析求解的问题,转化为一个等价的、可通过随机试验来估算的概率模型。
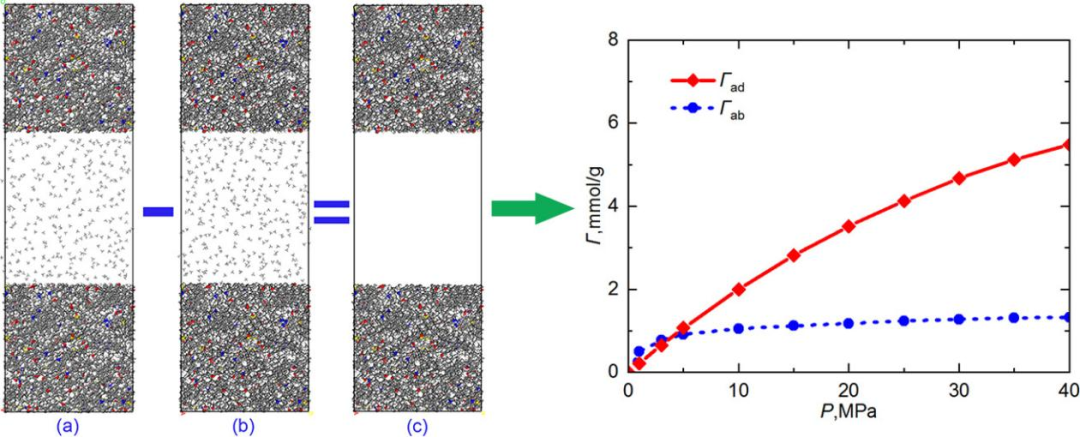

图1DOI: 10.1016/j.molliq.2020.114364
在材料与化学领域,蒙特卡洛模拟是一种强大的“计算实验”工具,它通过在原子或分子尺度上模拟体系的微观状态演化,来预测宏观热力学性质。
其理论基础根植于统计力学,核心在于如何有效地在庞大的构型空间中进行采样。这主要依赖于以下几个关键理论概念:
统计系综(Statistical Ensembles):模拟首先需要确定体系所处的宏观环境,这在统计力学中由“系综”来描述。研究者依据目标选取合适的系综,如固定粒子数、体积和温度的正则系综(NVT),固定粒子数、压力和温度的等温等压系综(NPT),或固定化学势、体积和温度的巨正则系综(μVT)。
每种系综都对应一个特定的概率分布函数,最常见的是玻尔兹曼分布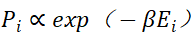 ,其中Ei是构型i的能量,
,其中Ei是构型i的能量,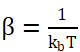 。
。
重要性采样(Importance Sampling):由于体系的绝大多数构型能量极高,对宏观性质的贡献微乎其微,因此均匀随机采样效率极低。重要性采样技术旨在优先对那些对体系性质贡献大的构型(通常是能量较低的构型)进行抽样,从而显著提高计算效率。
Metropolis算法(Metropolis Algorithm):这是实现重要性采样的最经典和基础的算法,由Metropolis等人在1953年提出。算法通过一个巧妙的接受/拒绝准则,引导体系的构型演化最终达到平衡态,并生成一个符合玻尔兹曼分布的马尔可夫链。
蒙特卡洛模拟的适用范围
蒙特卡洛模拟不涉及牛顿运动方程的求解,不直接提供体系的时间演化信息,因此其主要优势在于高效地计算体系的平衡态性质。在材料和化学领域,其应用极为广泛,涵盖了从气体吸附到晶体生长的诸多现象。
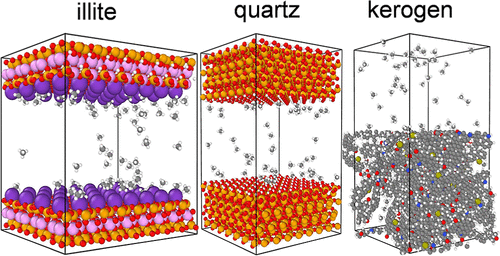
图2DOI: 10.1021/acs.energyfuels.2c03156
吸附等温线计算:这是MC模拟最成功的应用之一。通过在巨正则系综(GCMC)中模拟气体分子在多孔材料(如金属有机框架MOFs、沸石分子筛等)孔道内的吸附–脱附过程,可以精确预测材料在不同压力下的气体吸附量,从而绘制出吸附等温线。
模拟结果通常能与实验数据良好吻合,为筛选和设计新型储气、分离材料提供了关键理论依据。
相变预测:MC模拟能够有效地研究物质的相变行为。通过在模拟过程中系统地改变温度或压力等参数,可以观察到体系宏观性质(如能量、密度、序参数)的突变,从而确定相变点(如熔点、沸点、临界点)。这对于理解材料的相图、预测新相的稳定性具有重要意义。
聚合物构象与溶液性质:对于链状大分子,其构象数量极其庞大。MC模拟,特别是利用构象偏倚(Configurational Bias Monte Carlo, CBMC)等算法,能够高效地采样聚合物链的构象,用于研究其回旋半径、末端距等尺寸性质,以及聚合物在溶液或熔体中的相行为。
怎么做蒙特卡洛模拟
定义模型与参数
首先需要建立体系的原子或分子模型,包括定义模拟盒子的大小、形状和边界条件(通常为周期性边界条件)。然后,最关键的一步是选择描述粒子间相互作用的力场(或势函数),如Lennard-Jones势、Coulomb势等,并确定其参数。力场的准确性直接决定了模拟结果的可靠性。
选择模拟软件与系综
根据研究问题选择合适的模拟软件包。通用分子模拟软件如LAMMPS等包含MC功能。此外,还有专门用于MC模拟的软件,如RASPA、Cassandra以及用于晶体热力学计算的CASM/CASINO。同时,根据研究目的选择合适的统计系综(如NVT, NPT, GCMC)。
设置模拟参数与执行
设置模拟的宏观条件,如温度、压力、化学势等。定义MC的尝试移动类型(move set),如单个粒子的平移、旋转,分子的插入/删除,或者模拟盒子的体积变化等。设置模拟的总步数,其中一部分用于体系达到平衡(弛豫阶段),另一部分用于统计采样(生产阶段)。
平衡判断与数据分析
在模拟过程中,需要监测体系能量、密度等关键物理量,判断体系是否达到平衡。通常,当这些物理量在某个平均值附近稳定波动时,可以认为体系已达到平衡。平衡后,继续运行模拟并记录每个采样步的构型和物理量。
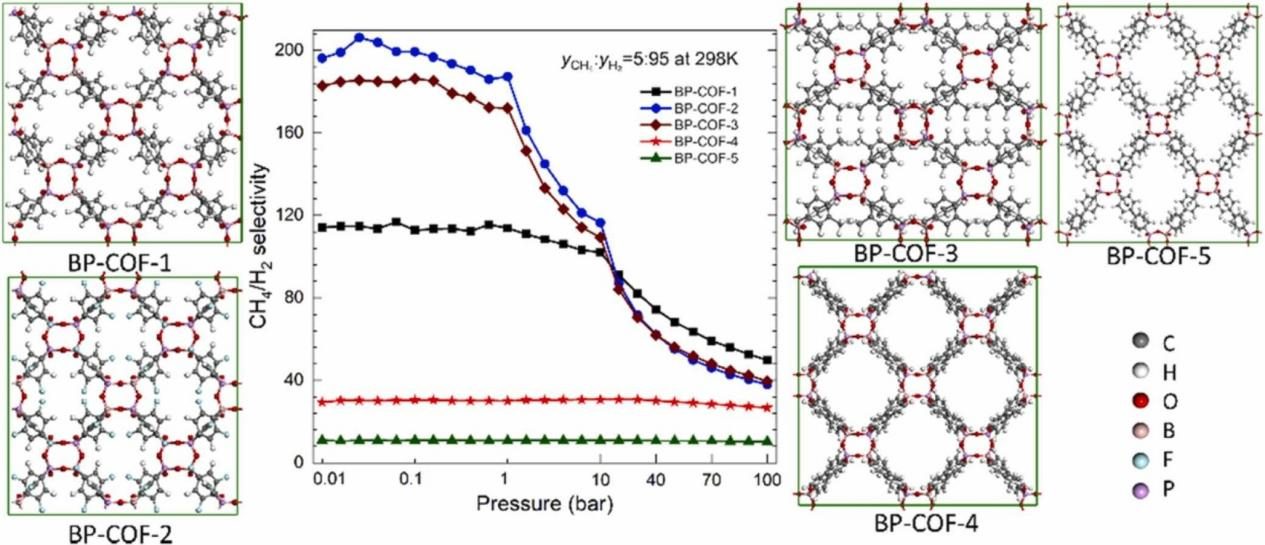
图3DOI: 10.1016/j.mtcomm.2022.104374
统计分析与结果解读
小结