

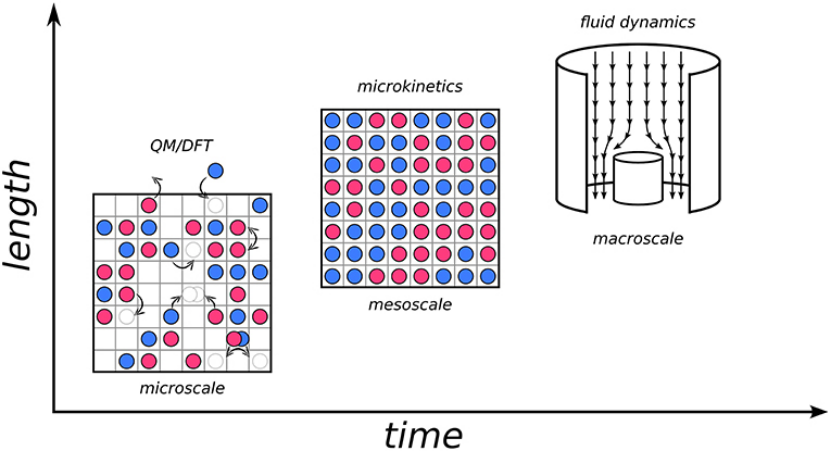
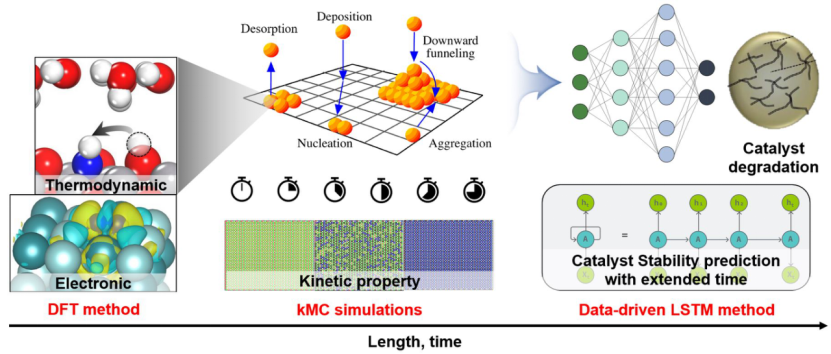
蒙特卡洛动力学模拟(Kinetic Monte Carlo, kMC)是一种基于随机过程的数值模拟方法,广泛应用于物理、化学、材料科学等领域,用于研究复杂系统的动态演化过程。以下将从基本原理、应用案例、示意图解析及与其他方法的对比等方面展开详细论述。
基本原理与核心框架
蒙特卡洛动力学模拟的起源可追溯至20世纪40年代的曼哈顿计划,最初用于中子扩散研究。其核心思想是通过随机数生成系统状态转移路径,并结合时间信息模拟动态过程。与传统蒙特卡洛方法(如Metropolis算法)仅关注平衡态性质不同,kMC能够模拟系统的时变演化,尤其适用于罕见事件(如化学反应、扩散)的动力学研究。

https://doi.org/10.3389/fchem.2019.00202
粗粒化与状态空间
kMC的核心在于对系统的粗粒化建模。系统被抽象为一系列长期稳定状态(long-time states),状态间的转换由激活能(activation energy)和速率常数(rate constants)决定。
例如,在表面催化反应中,吸附分子在晶格位点间的迁移即被视为状态间的转换。通过忽略快速过程(如高频振动),kMC能够跨越较大的时间尺度(从微秒到秒级),显著提高计算效率。
主方程与马尔可夫链
系统的演化通过主方程(Master Equation)描述,其形式为:
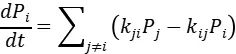
其中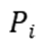 为状态的概率,
为状态的概率,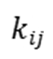 为状态到
为状态到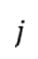 的速率常数。kMC算法通过构建马尔可夫链,按指数分布随机选择下一个状态转移事件,并累积时间步长。这一过程要求满足微观可逆性(detailed balance),即平衡态下正向与逆向速率满足玻尔兹曼关系。
的速率常数。kMC算法通过构建马尔可夫链,按指数分布随机选择下一个状态转移事件,并累积时间步长。这一过程要求满足微观可逆性(detailed balance),即平衡态下正向与逆向速率满足玻尔兹曼关系。
算法流程
典型的kMC流程包括以下步骤:
1.初始化:确定初始状态及所有可能的转移路径与速率。
2.事件选择:根据速率权重随机选择下一个事件。
3.时间推进:根据总速率生成时间增量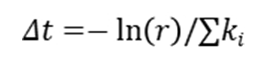 (
(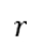 为随机数)。
为随机数)。
4.状态更新:执行选定事件,更新系统状态。
5.循环迭代:重复步骤2-4直至达到预设时间或状态。
应用案例与文献分析
kMC方法在多个领域展现了强大的适用性,以下结合具体文献展开说明。
表面催化与化学反应
Savva等(2023)利用kMC研究了CO氧化、N₂O分解等表面反应的动力学过程。例如,在CO氧化反应中,kMC模拟揭示了催化剂表面吸附位点的覆盖度对反应速率的影响。
通过构建晶格模型,研究者追踪了CO和O₂分子的吸附、扩散及反应路径,发现低温下O₂的解离吸附是速率决定步骤。文献中的示意图常展示晶格快照(如O*、CO*的分布),直观反映不同时间点的表面覆盖状态。
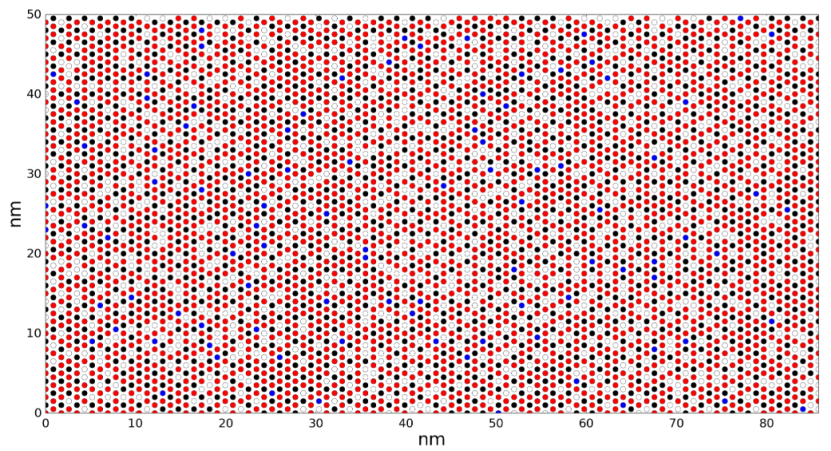
https://doi.org/10.1016/j.cattod.2021.03.010
Sharma等(2023)将kMC应用于锂离子电池的枝晶生长模拟。通过建模锂离子在电极表面的扩散与沉积,研究发现局部电流密度不均匀性会引发枝晶形核。kMC轨迹图显示,枝晶尖端的高曲率区域因电场集中而加速锂沉积,这一结果与实验观测吻合。例如,下图展示了不同扫描速率下的循环伏安曲线,积分结果进一步量化了晶格占有率随电位的变化,揭示了滞后效应的微观机制。
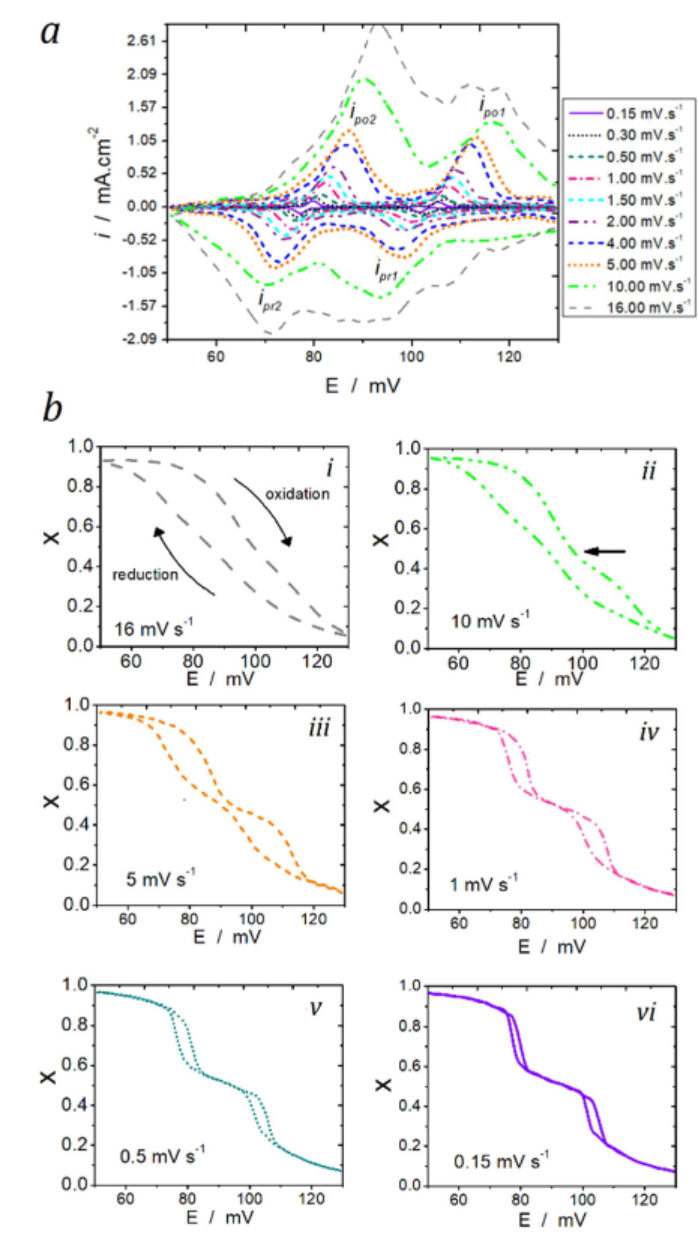
https://doi.org/10.1016/j.electacta.2019.135439
聚合物流变学
Mavrantzas(2021)通过kMC模拟研究了聚合物熔体的非平衡流变行为。通过引入合成场(synthetic field)描述剪切作用,模拟捕捉到链段取向与黏弹性响应。研究中的轨迹图显示,长链聚合物在剪切场下呈现明显的各向异性分布,与宏观流变模型预测一致。
蛋白质–表面相互作用
Ozboyaci等(2016)结合kMC与布朗动力学(BD)模拟了蛋白质在材料表面的吸附过程。kMC用于采样蛋白质的构象空间,而BD模拟扩散驱动的结合路径。结果显示,静电相互作用主导了初始吸附取向,而疏水作用影响长期稳定性。此类研究常通过势能面(Potential Energy Surface, PES)图展示不同吸附构型的能量差异。
KMC模拟中的示意图与解析
kMC论文中常见的示意图类型包括势能面、轨迹图、晶格快照及动态过程曲线,以下结合具体文献详述。
势能面与状态转换
下图(MMonCa, 2013)展示了状态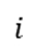 和之间的能量差(
和之间的能量差(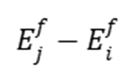 )及势垒
)及势垒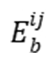 。蓝色球体表示系统在状态的稳定位形,红色箭头表示跨越势垒的转换路径。此类图直观解释了kMC中事件选择的概率权重,即速率常数
。蓝色球体表示系统在状态的稳定位形,红色箭头表示跨越势垒的转换路径。此类图直观解释了kMC中事件选择的概率权重,即速率常数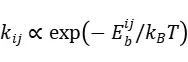 。
。
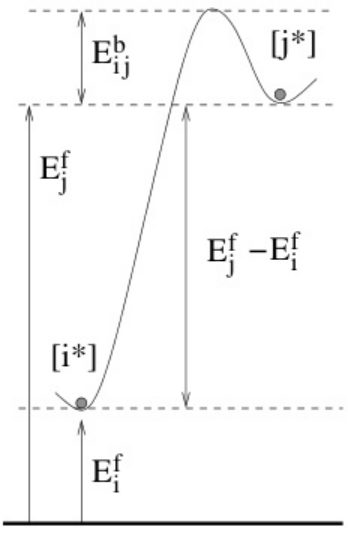
https://doi.org/10.1016/j.cpc.2013.07.011
粗粒化过程示意
Kaiser等(2018)展示了系统动力学的粗粒化过程。图中能量曲线上的“长期状态”对应系统的亚稳态,虚线表示快速振动等被忽略的过程。图(b)则描绘了从初始状态到多个终态的可能路径,反映kMC算法中事件树的随机分支特性。
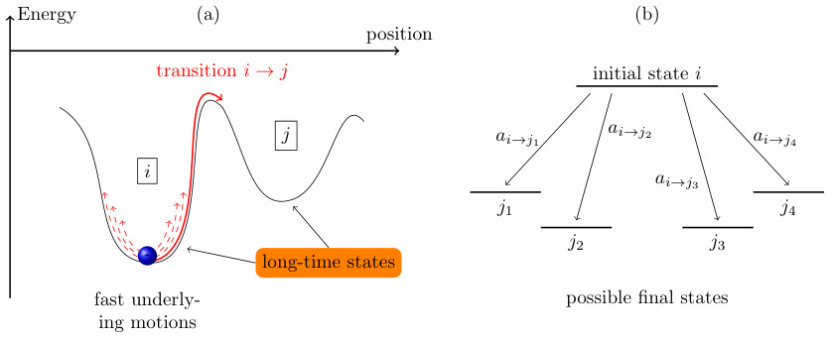
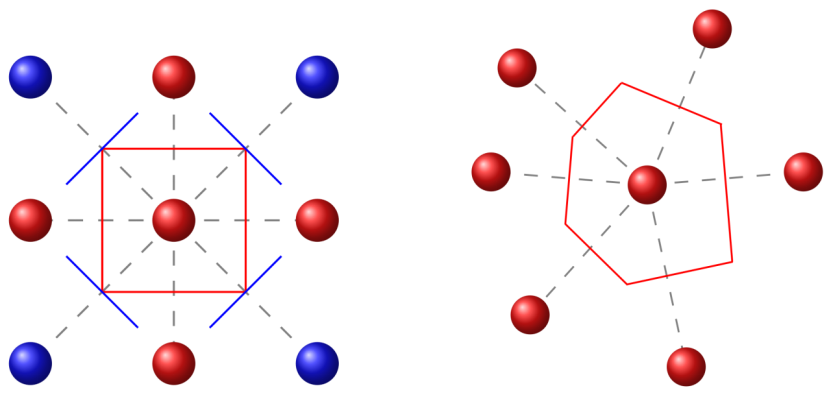
https://doi.org/10.3390/a11040037
动态轨迹与时间演化
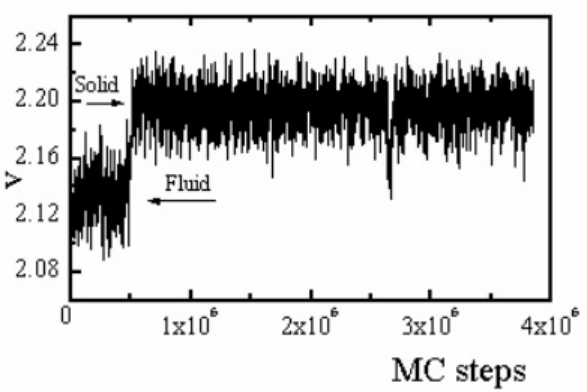
Jagla(1998)的MC步骤–变量V曲线展示了系统从非平衡态向平衡态的演化。早期阶段V剧烈波动(对应快速弛豫),随后趋于稳定(进入准平衡态)。约3×10⁶步时V的骤降可能源于罕见事件(如晶格重构),随后恢复稳定,印证kMC对长时间尺度的捕捉能力。
晶格快照与稳态分析
Li等(2020)的晶格快照显示了O*、CO*在催化剂表面的分布。红色与黑色圆圈分别代表O和CO吸附物种,背景色标可能表示局部覆盖度或能量。此类图用于验证横向相互作用(如吸附物-吸附物排斥)对反应动力学的修正效应。
有研究者开发了一种KMC 模拟方法,用于 TMD 范德华外延生长建模。该KMC 方法引入了自下而上合成中 TMD 的完整材料参数:金属和硫属元素在基底和生长的 TMD 表面的吸附/解吸/扩散、TMD 堆垛顺序、硫属元素/金属比、薄片边缘扩散和空位扩散。
KMC过程呈现出多种动力学行为,这与实验中观察到的各种生长行为相关。KMC方法可用于研究和预测生长机制,为指导实验研究提供定性建议。
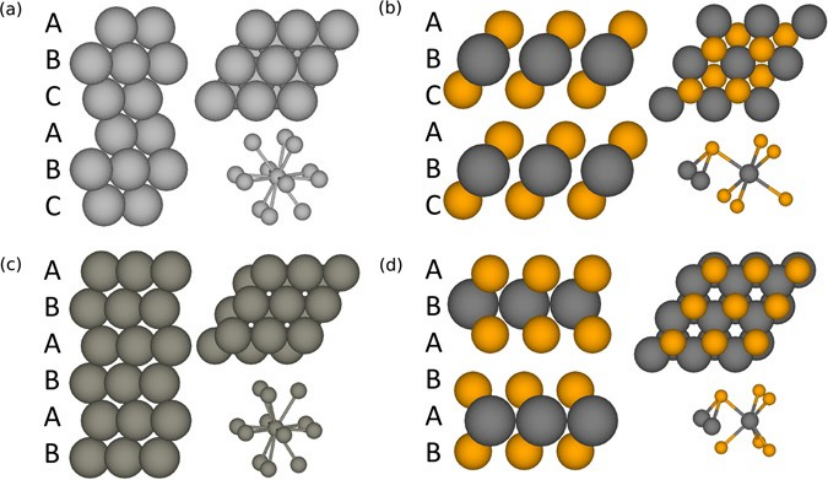
https://doi.org/10.1038/s41598-017-02919-2
KMC与分子动力学的对比
kMC与MD是互补的模拟方法,主要差异体现在时间尺度、计算效率及适用场景。
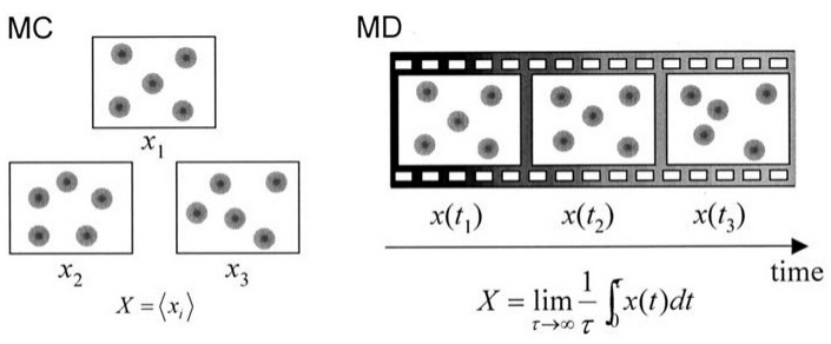
时间尺度与粗粒化
MD通过求解牛顿方程追踪原子轨迹,时间步长通常在飞秒级,难以模拟秒级过程。kMC通过粗粒化跳过快速振动,直接模拟状态转换,适用于稀有事件(如扩散、反应)。例如,金属中空位迁移的激活能较高,MD需数亿步才能观测到一次跃迁,而kMC可高效模拟此类过程。
计算效率与精度
MD的精度依赖于势函数和积分算法,但计算成本随体系规模立方增长。kMC的计算量取决于状态数与事件列表,适合中等规模系统。例如,Chen(2018)对比发现,kMC模拟表面反应的效率比MD高2-3个数量级,但需预先确定所有可能的转移路径。
混合模拟方法
Markvoort(2014)提出MD-kMC混合算法,MD用于短时精确计算局部动力学,kMC处理长时全局演化。例如,在纳米通道气体输运中,MD模拟近壁区分子碰撞,kMC模拟主体流动,兼顾精度与效率。
总结与展望
蒙特卡洛动力学模拟通过粗粒化与随机事件采样,在介观尺度上高效解析复杂系统的动态行为。其在催化、能源材料、生物分子等领域的成功应用,彰显了方法的普适性。未来,结合机器学习(如加速事件列表生成)与量子计算(如优化随机数生成),kMC有望进一步突破时间与空间尺度的限制,为多尺度建模提供更强大的工具。
通过上述分析可见,kMC不仅是一种数值工具,更是连接微观机制与宏观现象的重要桥梁。其示意图的多样化与动态可视化(如轨迹、势能面)为理论结果的直观展示提供了丰富手段,而与其他方法的对比则凸显了其在特定场景下的独特优势。