

反应位点指的是化学反应中参与反应的原子或原子团,它们通常具有独特的电子或几何特征。例如,催化剂表面的金属原子、分子中的特定官能团、酶的活性中心等,都是典型的反应位点。不同的反应位点能影响反应路径、产物选择性以及催化效率。
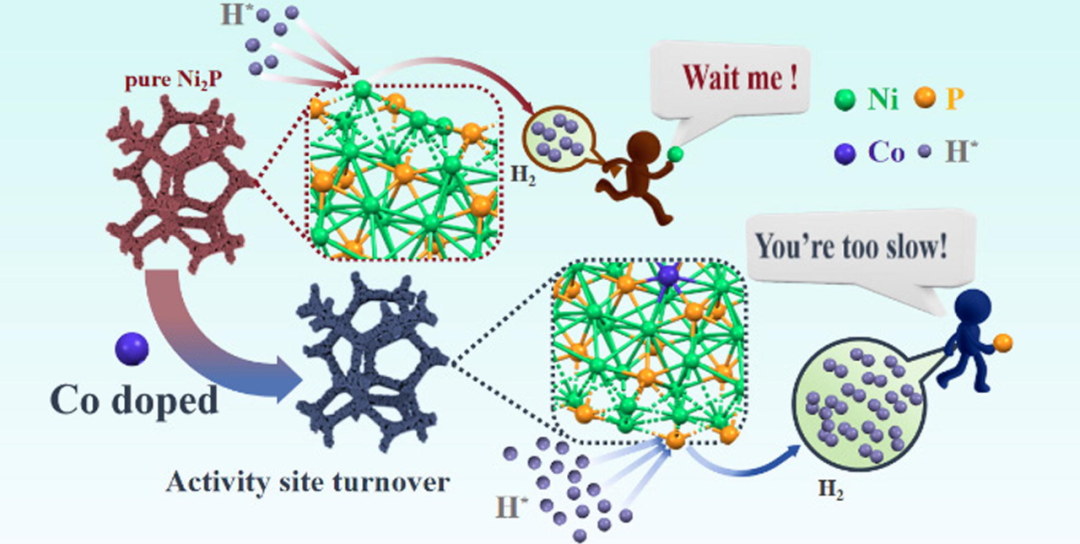

DOI: 10.1016/j.jechem.2024.02.006
反应位点的活性受以下几个因素影响:
电子结构:电子密度分布和能级匹配会决定反应位点的反应性。例如,金属催化剂表面的低配位金属原子通常比高配位原子具有更强的反应性,因为低配位原子具有较高的电子密度和较低的能量状态。
几何结构:反应位点的空间位置和形状也对其活性有重要影响。催化剂表面的台阶、边界或缺陷位置通常是活性位点。特别是在多晶面催化反应中,表面不同晶面暴露的原子会影响其吸附和反应的选择性。
微环境:溶剂、电解质、温度等环境因素也会影响反应位点的表现。温度和溶剂种类会改变反应物的解离和吸附方式,尤其在电催化和生物催化过程中,电场和pH值的变化也会显著影响反应位点的活性和稳定性。
通过理论计算,我们可以预测反应位点的活性、稳定性和选择性,甚至能够指导实验研究。常用的计算方法包括:
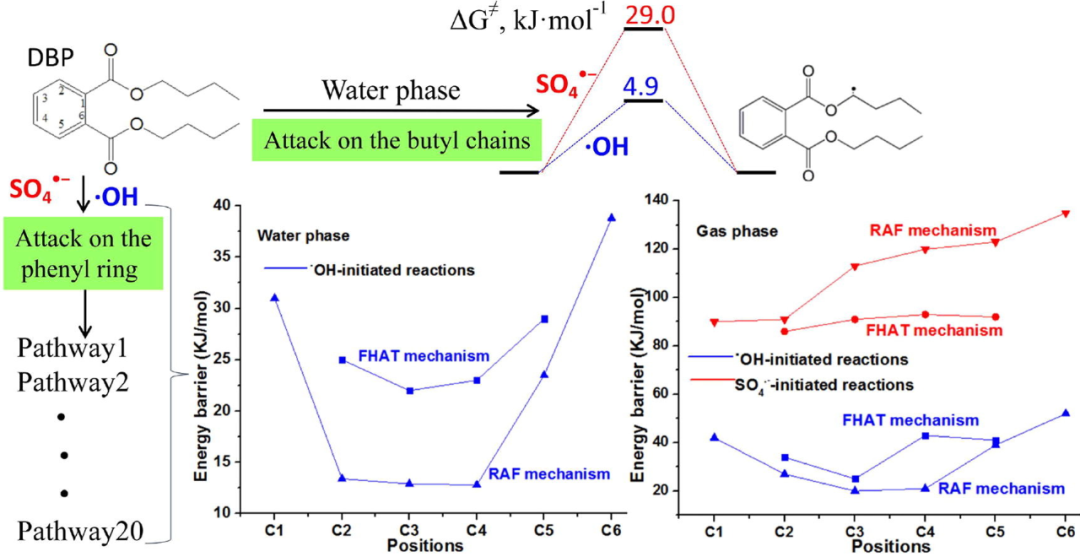
DOI: 10.1016/j.cej.2019.122680
密度泛函理论(DFT):密度泛函理论是研究反应位点电子结构和能量变化的主要工具。通过DFT计算,可以准确获得反应物、过渡态和产物的吸附能和反应能垒,从而预测反应路径和活性位点。例如,在催化反应中,DFT可以帮助我们判断催化剂表面哪部分原子或结构最适合吸附反应物,进而进行反应。
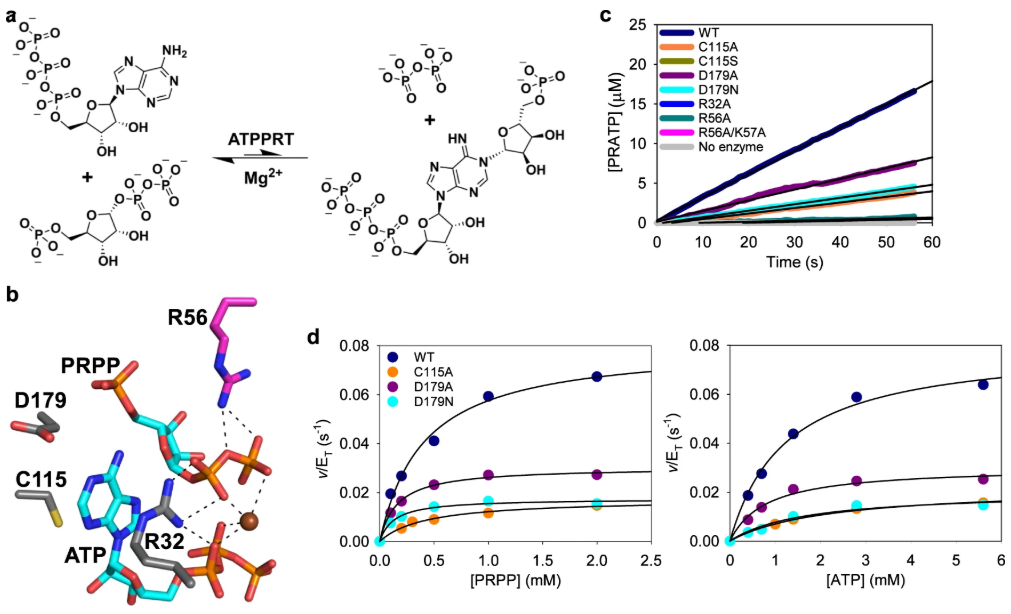
DOI: 10.1038/s41467-022-34960-9
分子动力学(MD)模拟有助于我们理解反应位点在不同温度、溶剂和电解质环境中的稳定性及其动态行为。例如,MD模拟表明,HisZ的结合限制了HisG S的动力学,支持了预先组织的活动位点,其中Arg56和Arg32在稳定WT-HisG S中的离去基团时起到了关键作用。在Arg56Ala-HisG S突变体中,HisZ通过调节Arg32的动力学部分补偿了Arg56的缺失。这些结果揭示了远程蛋白质–蛋白质相互作用如何通过恢复活性位点的静电预组织来增强催化弹性。
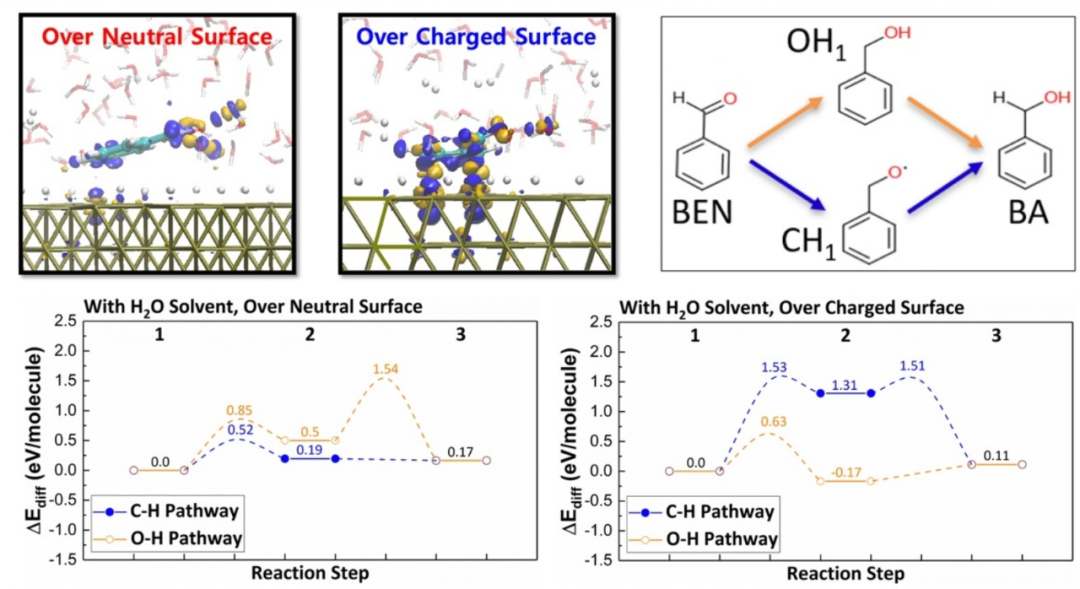
DOI: 10.1016/j.cattod.2020.07.039
从头算分子动力学模型(AIMD)结合了量子化学和经典动力学的方法,能够对反应机理和催化性能进行定量评估。通过密度泛函理论(DFT)计算得到的反应能垒和吸附能,可以构建反应速率方程,进一步分析反应位点的反应速率、选择性以及催化过程中速率控制步骤。该模型不仅帮助我们理解不同反应位点条件下反应的具体机理,还为催化反应的优化提供了重要的理论支持,能够为实际催化过程的调控和改进提供科学依据。
反应位点是化学反应中最易发生反应的特定位置,其活性受电子结构、几何构型和微环境等因素影响。通过密度泛函理论(DFT)、分子动力学(MD)等计算方法,可以预测反应位点的活性与稳定性。通过深入理解反应位点,可以显著提升催化效率和选择性,为能源转换、绿色合成等领域的技术进步提供支持。