

弱相互作用分析是计算化学中研究分子间非共价键作用的关键技术,主要包括氢键、π-π堆积、范德华力等类型。
通过约化密度梯度(RDG)分析可视化相互作用区域,蓝色凹陷对应氢键,绿色区域表征范德华力;电子密度分析可量化键临界点(BCP)的ρ值和拉普拉斯量,区分共价键(∇²ρ∇²ρ>0);
自然键轨道(NBO)分析通过二阶微扰能E(2)评估超共轭效应;Hirshfeld表面分析则通过dnorm参数和形状指数识别分子间接触特征。这些方法在药物设计、材料科学等领域具有重要应用价值。
弱相互作用分析是计算化学中研究分子间非共价作用的核心手段,这些看似微弱的力(1-10 kcal/mol)在分子识别、自组装和生物大分子构象稳定中具有决定性作用。
以下华算科技从分析指标体系、物理意义及可视化表征三个维度展开系统阐述,并配以典型分析图示说明。
弱相互作用的类型
弱相互作用主要包括氢键(如O-H···O)、π-π堆积(苯环堆叠)、CH-π相互作用(甲烷与苯环间)及范德华力(瞬时偶极作用)。
虽然单个作用能仅为共价键的1/10-1/100,但协同效应使其在DNA双螺旋稳定(氢键网络)、蛋白质折叠(疏水作用)和MOF材料孔隙调控(π-π定向组装)中不可或缺。例如,药物分子与靶标蛋白的结合常依赖多重弱相互作用的协同效应。
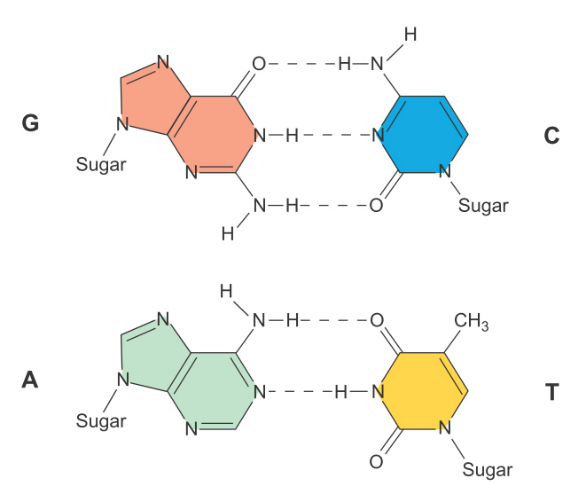

DNA双螺旋结构中的氢键网络示意图
核心分析指标及其意义
约化密度梯度(RDG)
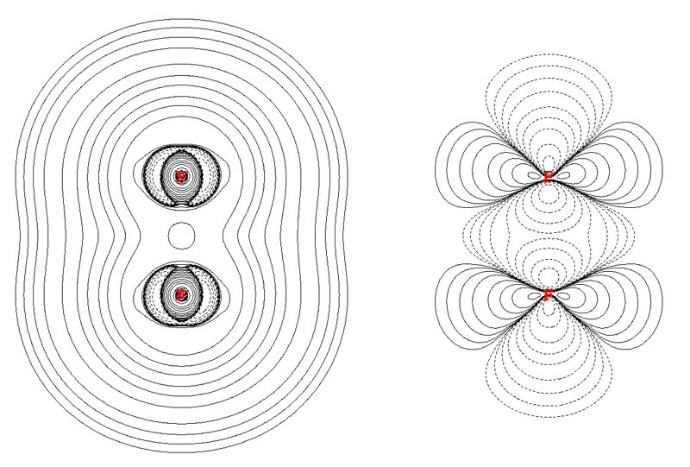
RDG通过电子密度梯度归一化处理实现弱相互作用可视化。其物理意义体现在:
电子密度平缓区标记:RDG≈0的区域对应相互作用位点,如氢键在RDG等值面图中表现为蓝色凹陷。
作用类型区分:结合sign(λ₂)ρ参数,蓝色区域(sign(λ₂)ρ₂)ρ≈0)为范德华力。
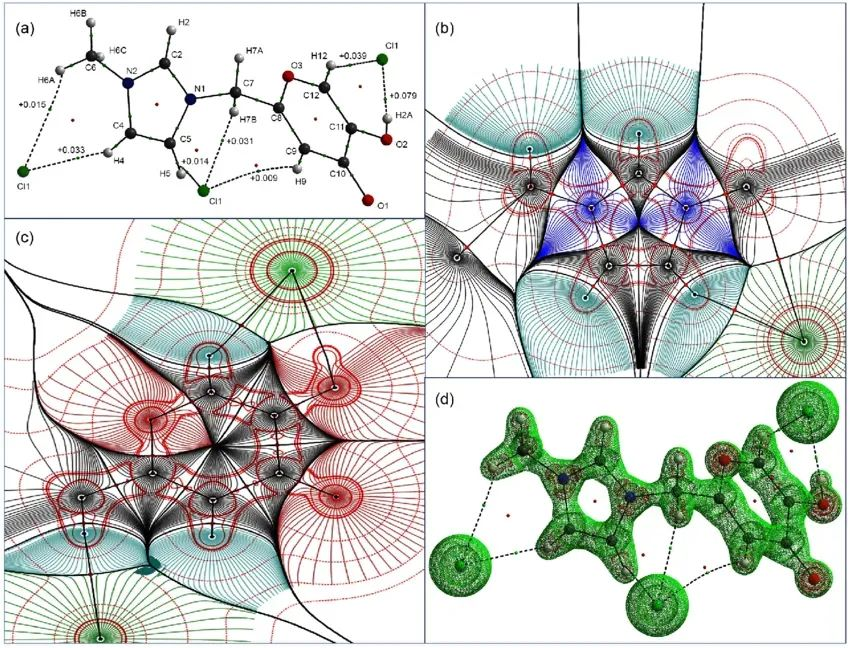
DOI:10.5935/0103-5053.20140220
电子密度分布(ρ(r))
通过量子化学计算获得的ρ(r)三维函数揭示:
键临界点(BCP)分析:电子密度在键路径极大值点(∇ρ=0)的数值ρ(BCP)可量化作用强度,如氢键的ρ(BCP)通常为0.002-0.035 a.u.。
拉普拉斯量(∇²ρ):共价键区域∇²ρ∇²ρ>0(电子离域),如图1B中H···O键的电子密度分布。
自然键轨道(NBO)分析
NBO将分子轨道转换为化学直观的定域轨道,其物理意义包括:
超共轭效应量化:通过二阶微扰能E(2)计算供体(如孤对电子)与受体(如σ轨道)的相互作用能。例如,甲醇中O孤对→C-Hσ的E(2)约为5 kcal/mol。
电荷转移分析:NBO电荷分布可识别关键原子(如氢键供体氧的电荷偏移),图中展示ATP分子中磷酸基团的电荷转移路径。
DOI: 10.3866/PKU.DXHX201603010
基于原子范德华半径划分的分子表面,通过两个核心参数:
dnorm:归一化接触距离,负值(红色)表示接触距离小于范德华半径和,对应强相互作用。
形状指数:曲率参数揭示表面凹凸特征,如π-π堆积表现为互补的”凸-凹”模式。

DOI:10.1016/j.molstruc.2025.142608
相互作用区域指示符(IRI)
IRI通过电子密度Hessian矩阵特征值(λ₁,λ₂,λ₃)构建,其优势在于:
无需预设作用类型:IRI等值面可同时显示共价键(高压缩区域)和弱相互作用(低压缩区域),如图1E中苯二聚体的π-π堆积与C-H键对比。
灵敏度高:可检测能低至0.5 kcal/mol的作用,适用于复杂体系如蛋白质-配体界面 。
分析流程
以金属有机框架(MOF)材料设计为例:
RDG+IRI联用:先通过RDG筛选潜在作用位点,再用IRI细化作用类型(如区分π-π堆积与CH-π作用)。
NBO验证:计算金属节点与有机配体间的电荷转移能,验证配位键的共价成分。
Hirshfeld量化:统计晶体中各类接触的占比(如氢键占表面积百分比),关联孔隙率与稳定性。