

说明:本文华算科技介绍了界面工程的概念、核心基础(界面电子结构、化学自由能等)及界面结构构建方法(外延生长、异质包覆等),读者可系统学习到界面工程提升催化剂性能的机制,了解其通过调控电子、几何结构等优化催化活性与稳定性的关键原理及实践策略。
什么是界面工程
通常,电化学反应发生在固体催化剂的表面或界面。为在特定催化条件下获得更优性能,精心设计的电催化剂需具备优化的物理或化学性质,以同时促进吸附和脱附过程。
在各类电催化剂设计策略中,界面工程是提升催化剂电催化性能的可行且有效策略之一。界面结构通常由两种或多种不同组分形成,理论上可作为不同组分间电子或中间体传输的通道。
通过合理调控界面原子排列,可调控电催化剂的物理化学性质,这对电催化性能具有重要影响。此外,不同组分间的强耦合作用及形成的界面不仅能提升电子导电性,还可增强电催化过程中的稳定性。
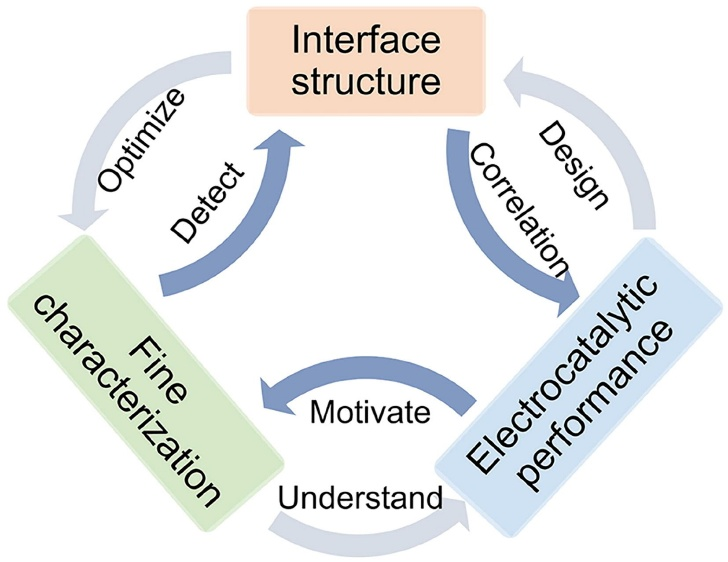

图1. 界面工程电催化剂的结构-表征-催化性能关系图。DOI: 10.1016/j.mattod.2021.02.004
界面工程基础
界面工程因其可通过独特的物理化学性质合理设计以提升电催化活性、稳定性和选择性,受到日益广泛的关注。
界面电子结构
电催化反应发生在催化剂表面或界面,过程涉及多电子转移及反应中间体的生成。电催化剂与反应物的相互作用对催化反应至关重要,而这种相互作用很大程度上取决于表面或界面的原子排列与电子结构。
界面工程可调控几何结构、晶体结构和电子结构,对电子、质子转移及反应物交换产生可调控的协同作用;界面结构上的电子重分布已被证实是促进电荷从催化剂向反应中间体转移的有效方式。
在各类分级电催化剂中,已构建出多种形式的界面。如图2所示,组分A和B之间形成界面,为电子和质子转移提供通道。
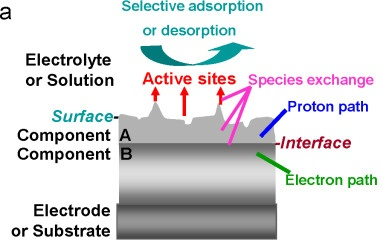
图2. (a)理想三相边界示意图:电子通道、质子通道与溶液相交汇于活性位点。DOI: 10.1016/S1872-2067(15)60911-1
通常,界面对电催化活性的作用机制包括两种情况(如图3所示):在情况I中,界面为电催化剂中两种组分(A和B)间的电子传输搭建桥梁,电催化反应主要发生在组分A上,反应速率受A与反应物间的结合能影响;通过电子相互作用可调控表面环境,平衡催化剂与反应物间的吸附和脱附强度,以实现最优电催化性能。
与情况I不同,界面相互作用的另一种情况(情况II)是不同组分负责不同的催化过程。如图3所示,组分A和B分别负责吸附和脱附过程,此时界面的作用是为反应中间体从A向B的转移提供通道。
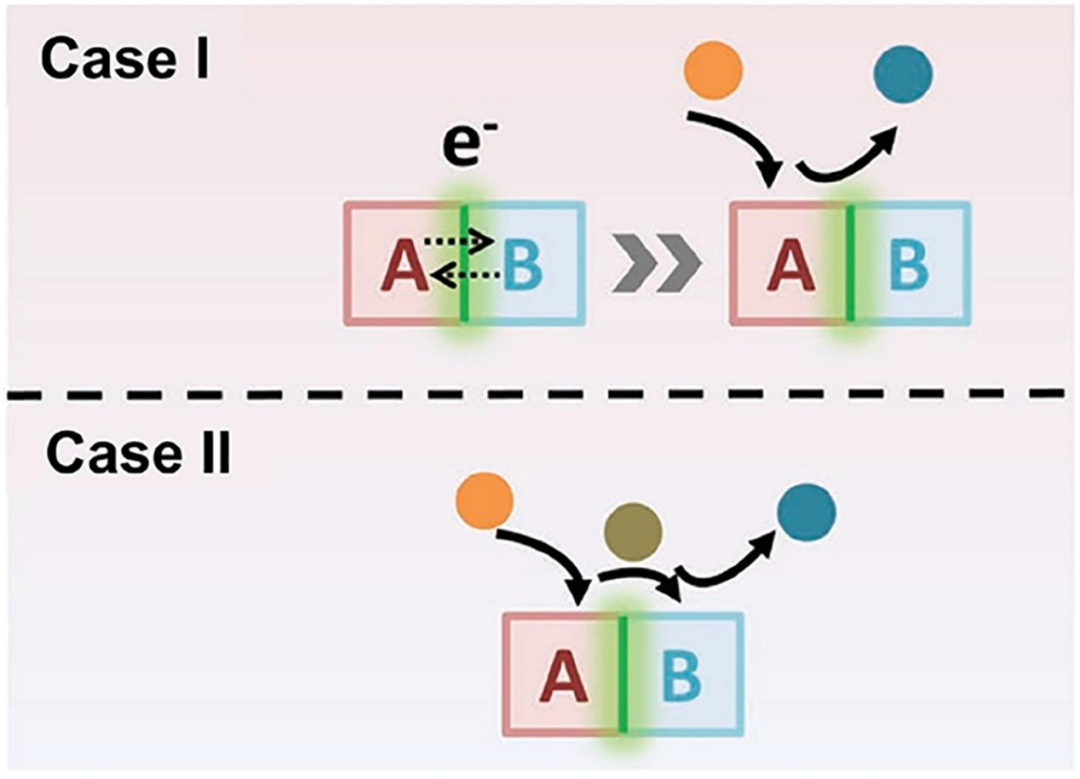
图3. A、B组分构成含界面催化剂:情形I,A提供活性位,B调控A表面环境以优化电催化;情形II,吸附与脱附分别发生在A、B。DOI: 10.1002/adfm.201806419
化学自由能
电催化反应的整体机制可总结为三个连续步骤:(1)反应物在电催化剂表面或界面的化学吸附;(2)反应物活化生成中间体;(3)最终产物从电催化剂脱附至电解液。
根据Sabatier原理,活性位点与反应物种的相互作用是影响电催化性能的关键因素,这种相互作用需强弱适中。因此,为促进整个电催化反应,需优化吸附和脱附步骤。
此外,d带中心能级越低,吸附质与金属的相互作用越弱—这是因为当金属催化剂表面发生吸附时,反键轨道会填充更多电子,导致电子从金属向吸附质的转移减少,进而产生弱结合作用和低活性。
如图4所示,为阐明界面工程对催化剂化学吸附的影响,研究人员设计了一种Ni-Co双金属氢氧化物前驱体的两步煅烧策略,构建多孔纳米级NiO/NiCo₂O₄异质结构,将其用作HER和OER的双功能电催化剂。
NiO纳米片与NiCo₂O₄基体间的强结合形成了丰富的活性界面区域,X射线光电子能谱(XPS)结果证实该区域存在电荷重分布。DFT计算表明,界面处的Ni位点更易成为吸附位点;与纯NiO和NiCo₂O₄相比,OH⁻更倾向于吸附在NiO/NiCo₂O₄异质结构的(100)界面表面,因为该界面对氢氧根离子的化学吸附自由能(ΔGOH)更低。
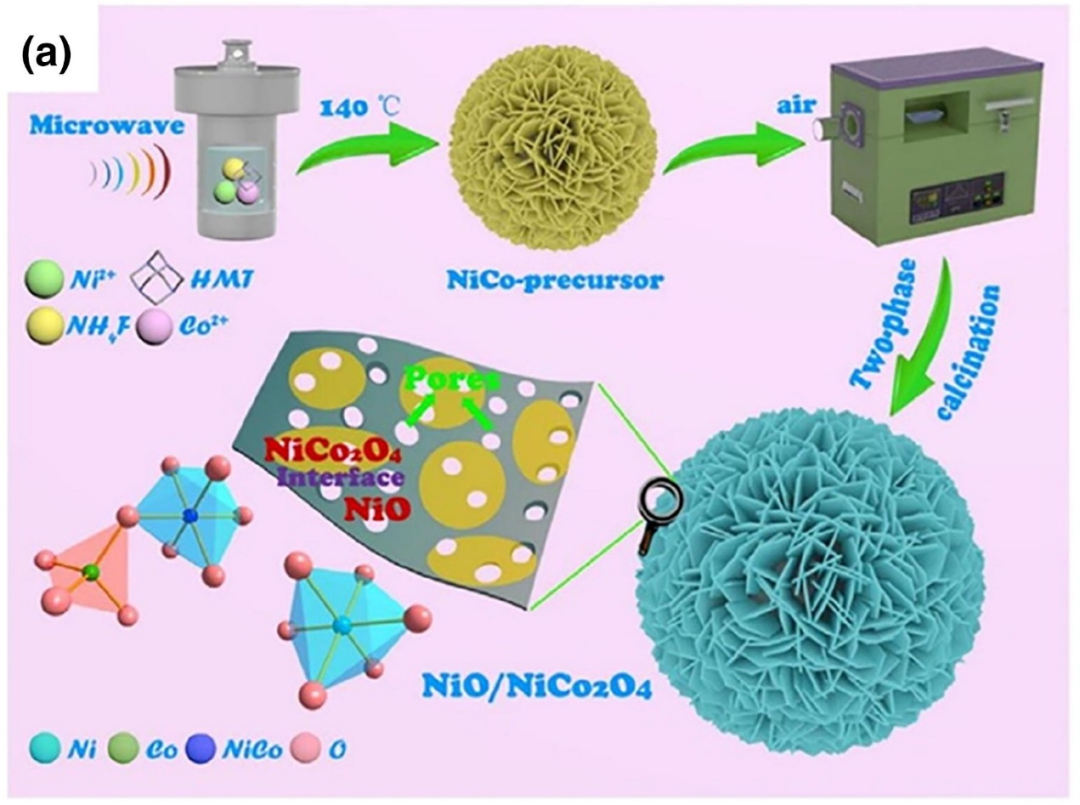
图4. (a)两步煅烧NiCo双金属前驱体制备多孔纳米NiO/NiCo₂O₄异质结构示意图,前驱体由微波辅助水热法合成。DOI: 10.1016/j.mattod.2021.02.004
功函数在理论上定义为0K时从材料中移除一个电子所需的最小能量,用Φ表示。材料表面的电荷重分布会产生表面偶极子,形成正负势能台阶,进而影响表面外的电子势能,最终有效改变功函数。
因此,可通过材料表面工程及改变电荷状态来调控功函数;从这个角度看,调控结构性质和局部几何构型是通过界面电荷重分布改变催化剂局部功函数的常用策略。功函数与电子从材料中移出的能垒相关,因此功函数越低,电子转移的能垒越低,更有利于电催化,功函数通常被用作评估电催化剂性能的关键参数。
紫外光电子能谱(UPS)对价带电子具有高灵敏度,已被广泛用于材料功函数的检测。研究表明,通过向碳基体中掺杂杂原子或引入其他化合物,可直接调控碳的电子能带结构,进而改变其功函数。
如图5所示,研究人员通过简单的配体交换反应结合热解过程,成功制备了嵌入氮磷共掺杂碳(N,P-C)中的焦磷酸钴(Co₂P₂O₇)纳米颗粒(NPs),形成具有丰富界面结构的纳米笼(Co₂P₂O₇@N,P-C)。
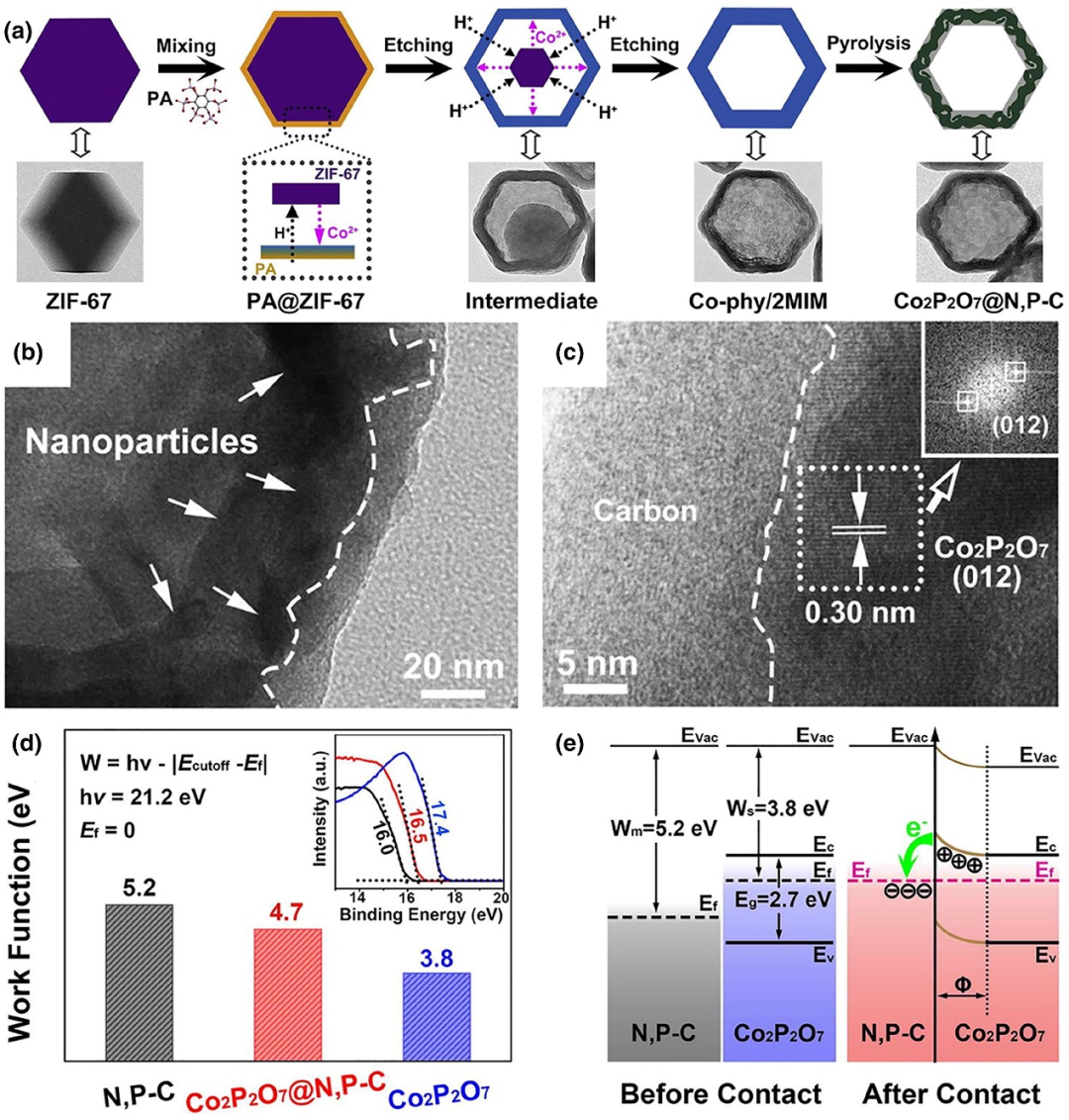
图5. (a)Co₂P₂O₇@N,P-C纳米笼的制备流程示意图。(b)Co₂P₂O₇@N,P-C纳米笼的透射电子显微镜(TEM)图像。(c)高分辨透射电子显微镜(HRTEM)图像。(d)Co₂P₂O₇、N,P-C及Co₂P₂O₇@N,P-C的功函数值(插图为对应的UPS光谱);(e)Co₂P₂O₇与N,P-C之间异质结的能带示意图(Evac=真空能级,Ec=导带,Ev=价带,Ef=费米能级,Eg=带隙,W=功函数,Φ=耗尽层)。DOI: 10.1016/j.apcatb.2019.118417
应变是描述固体材料形变的重要物理概念。应变效应通常形成于不同组分晶格参数不匹配的杂化材料中,并从界面向表面衰减。在特定材料中,可通过晶格畸变观察到应变效应引起的原子键长变化。内应变效应可调控表面电子结构,改变对吸附质的结合能,进而影响电催化剂的活性和稳定性。
d带小于半填充的早期过渡金属在晶格膨胀时表现出更低的吸附能,这与典型的晚期过渡金属情况相反。根据d带理论,膨胀应变会窄化d带宽度:对于早期过渡金属,d带窄化导致d带中心下移以维持d带填充度;对于晚期过渡金属,相同机制会使d带中心上移。
如图6所示,研究人员在氧化石墨烯(GO)负载的Pd-Au双金属纳米颗粒中,通过Pd与Au界面处的局部应变,提升了ORR的效率和稳定性。HRTEM结果显示,由于Pd与Au的晶格不匹配,Pd-Au界面附近的Pd外延层承受拉伸应力,而Au则承受压应力。
DFT计算表明,对于承受+5%拉伸应变的界面Pd位点,*O的结合能降低,而*OH的结合能增加。因此,速率决定步骤(O*+H₂O+e⁻→OH*+OH⁻)的能垒减小,ORR性能得到增强。
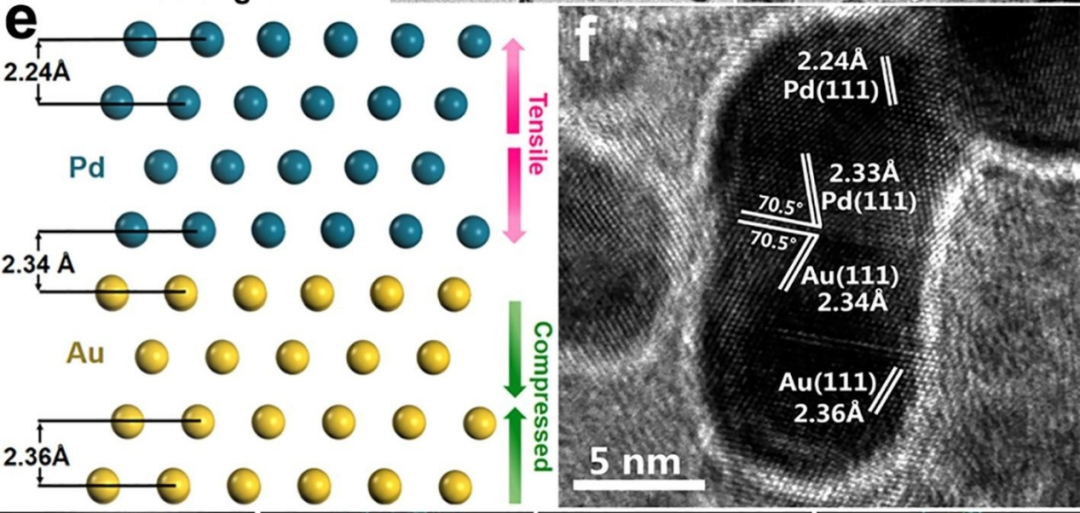
图6. (e)晶体结构示意图及(f)HRTEM图像,清晰展示Pd-Au晶格。DOI: 10.1016/j.cej.2020.124240
综上,电催化剂中两种不同组分形成的界面效应对催化活性和选择性具有重要影响:界面工程可调控电子结构,以优化电子和质子转移,平衡催化剂上中间体的吸附与脱附;此外,界面工程还可通过调控几何结构和化学吸附行为影响反应物在催化剂上的反应活性,而催化剂中的应变效应也能改变对吸附质的结合能,从而提升电催化活性、选择性和稳定性。
界面结构构建方法
外延生长
外延生长是原位构建具有目标晶相和暴露界面的杂化纳米结构的典型方法。两种晶体间的小晶格失配可最小化杂化纳米结构的界面能,这是实现外延生长的关键因素。溶液相外延和气相外延是制备具有不同结构和组分的杂化纳米结构的两种常用外延生长方法。
如图7所示,研究人员利用晶体结构的晶格匹配性,通过在H₂/Ar气氛下热处理,在暴露(001)晶面的二维三元硫代磷酸镍(NiPS₃)纳米片上原位外延生长二磷化二镍(Ni₂P)。
DFT计算表明,NiPS₃/Ni₂P外延界面处的内建电场可显著降低H吸附能垒,加速电子转移。实验测试结果进一步表明,所得NiPS₃/Ni₂P中的外延界面对HER和OER均有活性提升作用。
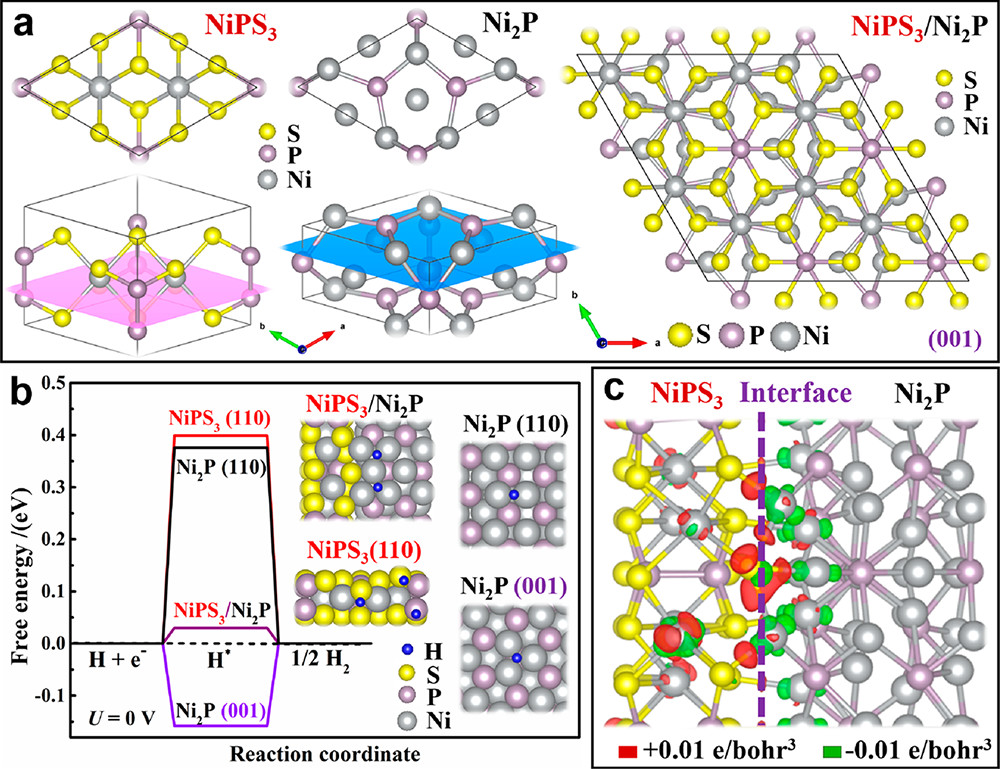
图7. (a)示意模型展示NiPS₃与Ni₂P之间的晶格匹配情况。(b)在平衡电位(U= 0V)下计算的NiPS₃/Ni₂P、Ni₂P(001)、Ni₂P(110)和NiPS₃(110)的ΔGH*,插图给出H*吸附的DFT优化构型。(c)NiPS₃/Ni₂P界面的电荷密度差分布,红色(绿色)分别表示电子积累(Δρ=+0.01e·bohr⁻³)和耗尽(Δρ=−0.01e·bohr⁻³);Ni、S、P、H原子分别以灰色、黄色、紫色、蓝色标示。DOI: 10.1021/acsnano.9b02510
异质包覆
异质包覆是在纳米尺度下构建核–壳结构中核与壳材料间界面活性位点的常用策略。核-壳结构可合理调控界面位点的电子/化学构型,提升催化效率和长期稳定性。
如图8所示,研究人员提出了一种简便的表面工程策略,通过在钴基金属有机框架(Co-MOFs)上包覆聚多巴胺(PDA)并辅助热解,合成ORR电催化剂-该催化剂中,Co纳米颗粒被多层氮掺杂石墨碳壳层包裹。
HRTEM图像清晰显示了Co纳米颗粒与碳层间的界面,XPS和N K边软X射线吸收近边结构(XANES)结果证实,此处形成了共价键合的Co-N-C界面结构,所制备的催化剂展现出理想的ORR性能。
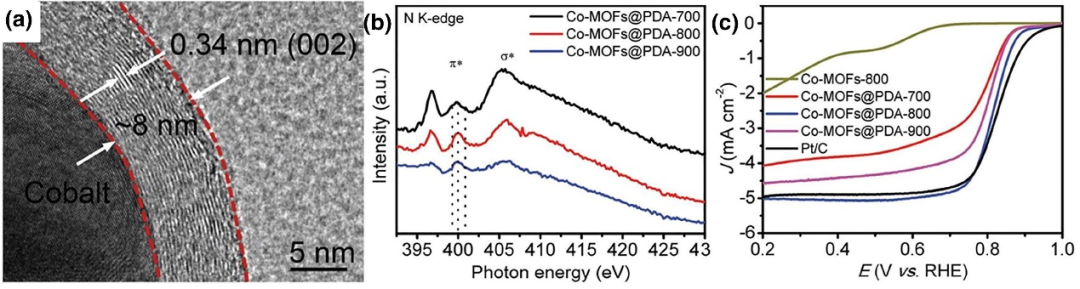
图8.(a)Co-MOFs@PDA-800的高分辨TEM图像。(b)Co-MOFs@PDA-n的N K边X射线吸收近边结构(XANES)。(c)各样品O₂饱和0.1M KOH溶液、1600rpm转速下的ORR线性扫描伏安(LSV)曲线。DOI: 10.1002/smll.201702074
杂化
两种纳米材料间的强杂化作用是电催化剂设计与合成领域的研究热点。金属氧化物、氮化物、硫化物以及碳基材料通过强化学键结合,被广泛用于杂化;其中,碳纳米管(CNTs)因导电性高且电化学稳定性优异,成为制备一系列杂化材料的热门选择。
如图9所示,研究人员利用最先进的钌(Ru)和氧化铱(IrOₓ)的高活性,通过分步多元醇策略合成了核-壳结构Ru@IrOx纳米晶,其界面处存在电荷重分布。
高角环形暗场扫描透射电子显微镜(HAADF-STEM)清晰识别出该纳米晶的界面结构—含Ir壳层(高衬度)和Ru核(低衬度)。得益于有利的价态和稳定的核-壳纳米结构,所制备的Ru@IrOx纳米晶在酸性环境中展现出优异的OER性能(活性高、稳定性好)。

图9. (E)沿[112]轴随机选取的Ru@IrOx核-壳二十面体纳米晶体的HAADF-STEM图像。(F)四方IrO₂与面心立方Ir分别沿[100]和[110]轴的投影结构模型。DOI: 10.1016/j.chempr.2018.11.010