

 同步辐射XAFS技术通过分析吸收原子周围的局部配位环境(如键长、配位数变化),能够灵敏地探测材料中的氧空位等缺陷。本文华算科技结合多个前沿案例,阐述了该技术在揭示空位形成机制、电子结构调控及其增强电催化性能等方面的关键作用。
同步辐射XAFS技术通过分析吸收原子周围的局部配位环境(如键长、配位数变化),能够灵敏地探测材料中的氧空位等缺陷。本文华算科技结合多个前沿案例,阐述了该技术在揭示空位形成机制、电子结构调控及其增强电催化性能等方面的关键作用。氧空位是指在特定的外部环境条件下,晶格结构中的氧原子会因能量变化等因素而脱离原有位置,从而引发氧的缺失,进而在晶格中产生氧空位现象。
在晶体结构中,氧空位是一种极为普遍的缺陷类型,它通常在晶体的制备阶段便已形成(如图1),也被称为固有缺陷。
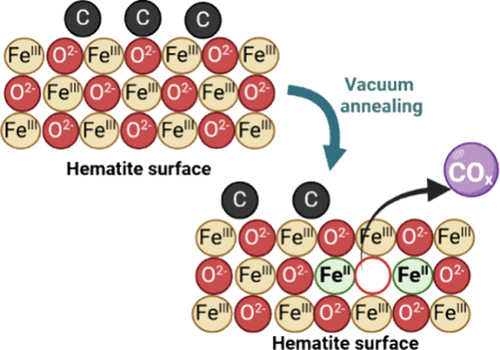
图1赤铁矿界面处碳介导的氧空位产生
(DOI: 10.1021/acs.jpcc.4c08423)
氧空位的形成可以通过多种方法实现,这些方法包括氢气热处理、离子掺杂、欠氧烧结、高温退火、机械研磨以及高能粒子轰击等。并且随着反应时间的延长,氧空位在材料中的相对含量往往会逐渐增加。
此外,通过在特定环境下进行高温热处理(如在真空、Ar、N2、He中),也可以在氧化物中引入氧空位。这些氧空位的形成不仅影响材料的电子结构和光学性质,还对材料的催化活性和能量存储性能产生重要影响。
在金属与绝缘体之间的相变过程中,它能够改变材料的电导率和电子结构。在磁阻效应方面,氧空位的存在可以调节材料的磁性,从而影响其对磁场的响应。
在催化领域,氧空位为反应物提供了活性位点,促进了化学反应的进行,提高了催化效率。在光吸收方面,氧空位能够改变材料对光的吸收特性,影响其在光电器件中的应用。
对于超级电容器的能量存储而言,氧空位有助于提高材料的电容性能,增强其能量存储能力。此外,在结构相变的形成过程中,氧空位也发挥着不可忽视的作用,它可能促使材料发生结构上的转变,进而影响材料的宏观性能。
空位作为复杂晶体缺陷,按缺失原子分阳离子、阴离子及混合空位。阳离子空位(如钛、铋、铜空位等),因阳离子原子量大、化学键强,需强烈物理/化学处理形成,难获高纯度单型,可调节电子结构,像TiO₂中钛空位能促电荷分离与表面反应;
阴离子空位(如氧、硫、氮空位等 ),原子量小、更易得稳定,氧空位1960年提出,2000年被发现可作反应位点,基于MvK机制,其及硫、氮等空位能增活性位点、改局部电子密度,如氮空位提升Fe掺杂C₃N₄中H₂O₂转换效率。
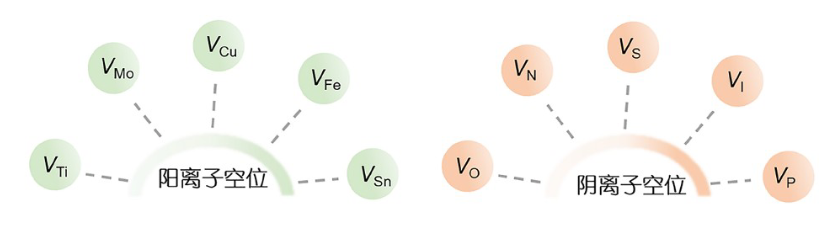
图2空位的分类
(DOI:10.1360/TB-2023-0609)
构建空位常见方法多样:热处理通过调控温度等影响空位形成,金属氧化物处理简单但耗时耗能;
元素掺杂灵活,可改变原子排列、平衡电荷,调掺杂条件能控空位浓度;
插层 – 剥离多用于二维层状材料,借层间作用与插层剂诱发空位;
表面化学蚀刻以酸碱等蚀刻剂产空位,常辅辅助技术,有优势也存蚀刻液问题;
还有β粒子辐射等其他方法,不同方法适用材料与问题有别,低碳环保且可控的制备方法仍待研究。
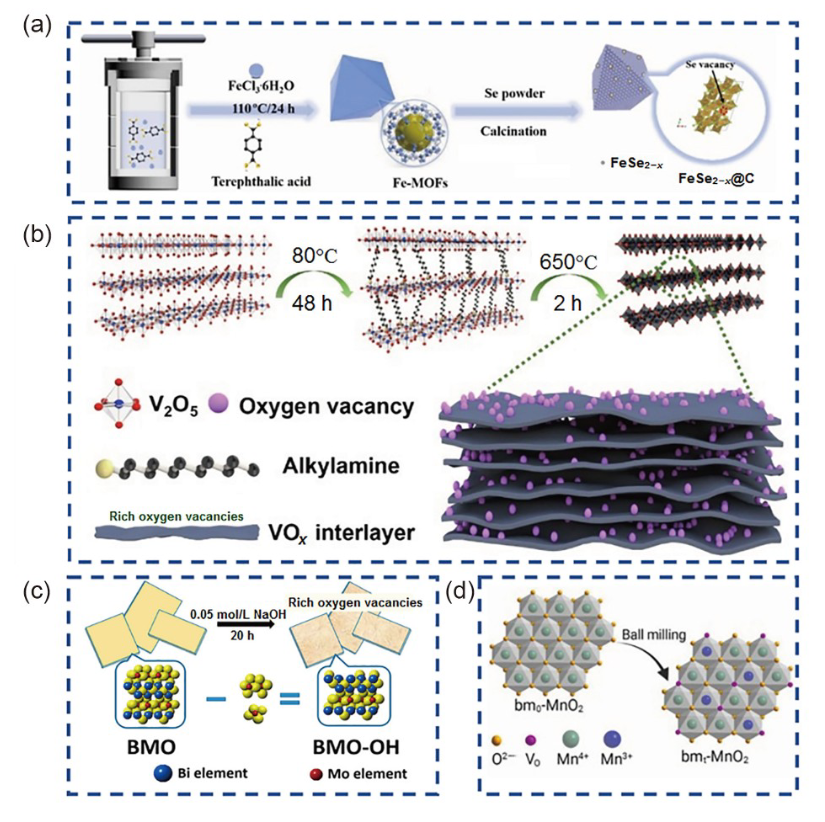
图3空位的构建方法:(a)热处理法构建FeSe2-x@C的Se空位,(b) 插层-剥离法构建V2O5中O空位,(c)化学蚀刻法构建Bi2MoO6中O空位,(d)球法构建MnO2中O空位。
(DOI:10.1360/TB-2023-0609)
X射线吸收精细结构(XAFS)能够精确地描述金属氧化物的局部结构和原子排列。其中,X射线吸收近边结构(XANES)光谱能够提供键长、配位数、原子种类和数量以及两原子之间的距离等详细信息。
对于低氧配位数的氧化物,其形成通常与氧空位的存在密切相关。通过XAFS观察相邻配位原子的距离及其峰值强度的变化,可以证实氧空位的存在。下面将介绍一些案例。
案例一:Advanced Science || XAFS检测镍合金中氧空位的产生

背景简介
FDCA(2,5-呋喃二甲酸)是高附加值生物质衍生物,可替代石油产品衍生物邻苯二甲酸生产可生物降解聚酯,减少污染和碳排放,且聚呋喃二甲酸乙二醇酯在气体阻隔性能上优于PET(聚对苯二甲酸乙二醇酯)。
目前,从HMFOR氧化合成FDCA是前景最好的方法,电催化相比热化学合成条件更温和。电催化活性主要受电极材料影响,常用过渡金属化合物等,需预氧化形成活性相。
金属氧化物虽稳定低毒,但导电性差,主要活性物质是NiOOH。引入氧空位可调电子结构,增强活性,常见方法有热处理、掺杂、机械力和激光烧蚀,其中激光加工灵活、高效,对电极表面影响小。
大连理工大学陶胜洋课题组通过紫外激光在商品化的合金表面引入氧空位,制备了含有氧空位的NiOOH电极(图8),氧空位可以调节电子结构,增强电子富集。
通过同步辐射XAFS,原位拉曼光谱和DFT理论计算揭示了Mo元素促进氧空位的形成过程,同时证明了氧空位有助于产生更多的NiOOH,提高电极对HMF的电催化活性。
图文解析
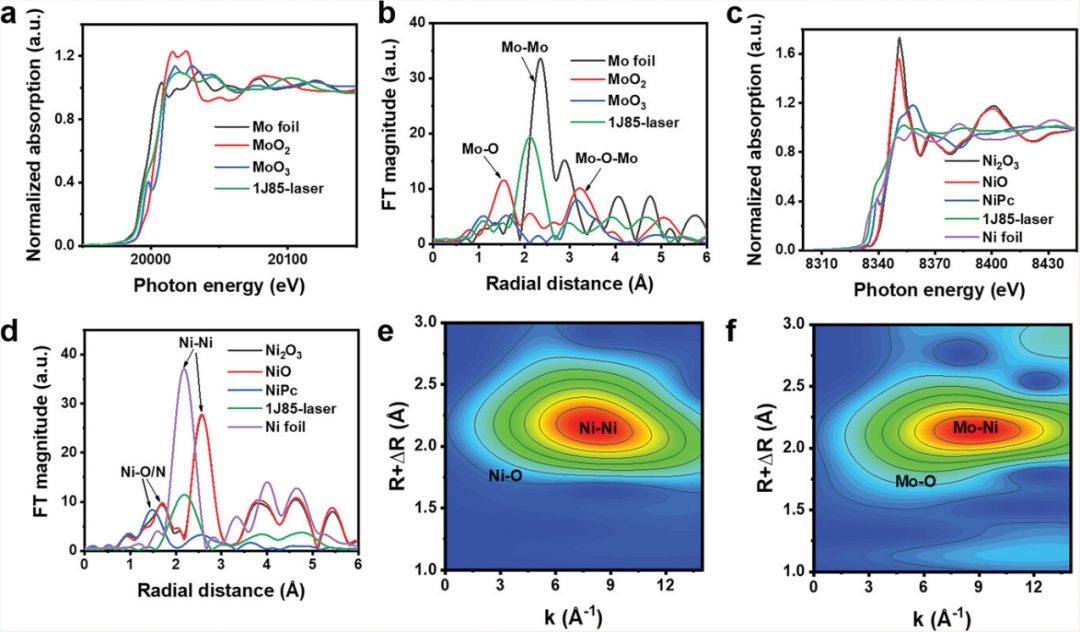
图4 a) 钼K边的XANES谱图。b) 钼K边EXAFS的FT曲线。c) 镍K边的XANES谱图。d) 镍K边EXAFS的FT曲线。e)和钼边f)的小波变换。
为了进一步研究1J85-激光电极中的原子配位和电子结构,进行了同步辐射X射线吸收精细结构(XAFS)测量。从钼K边的X射线吸收近边结构(XANES)(图8a)可以看出,1J85-激光中的钼价态高于钼箔而低于MoO₂。
扩展X射线吸收精细结构(EXAFS)被用来详细研究与钼相关的材料的配位结构。在钼K边的傅里叶变换(FT)EXAFS光谱(图8b)中,1J85-激光在2.2 Å附近有一个明显的振荡峰,位于Mo-Mo和Mo-O之间,这可能是由于样品中钼与其他金属的配位所致。
拟合结果表明,钼的配位峰主要由两条路径组成,即Mo-O和Mo-Ni。钼与镍的配位影响了镍的配位环境,这表明钼的存在可能促进了镍与氧的不饱和配位。
镍K边的XANES显示,1J85-激光中的镍价态介于0和+2之间(图8c),这表明镍与氧的不饱和配位导致了氧空位的产生。上述结果与XPS结论一致。在镍K边EXAFS的FT光谱(图8d)中,1J85-激光在2.2 Å附近有一个明显的振荡峰,这可能是Ni-Ni金属键。
与镍的氧化物相比,1J85-激光中Ni-Ni键的配位数减少,这是由于1J85-激光中镍的3d轨道填充度较低。较低的3d轨道填充度有利于电极与反应物的结合,从而增强电催化活性。根据拟合结果,镍配位峰的路径主要由Ni-O和Ni-Ni组成。
在小波变换EXAFS(WT-EXAFS)图(图8e,f)中,可以在R空间相应位置观察到金属原子的强烈WT信号。由于配位数较低,Ni/Mo-O在WT-EXAFS中的信号较弱。上述XAFS分析说明了1J85-激光电极中镍和钼原子的配位结构。
原文链接:
Junbo Liu, Shengyang Tao.Laser Promoting Oxygen Vacancies Generation in Alloy via Mo for HMF Electrochemical Oxidation
https://doi.org/10.1002/advs.202302641
DOI:10.1002/advs.202302641
案例二:ACS Catalysis|| 超薄且富含空位的CoAl层状双氢氧化物/石墨氧化物催化剂:在醇氧化中钴空位和氧空位的促进作用

背景简介
开发高效、经济的非贵金属催化剂以替代传统的贵金属催化剂,用于伯醇的选择性氧化反应。尽管贵金属催化剂在该反应中表现出色,但其高昂的成本和稀缺的资源限制了其广泛应用。
Co基催化剂因其良好的氧化性能而备受关注,但存在Co物种分散性差和TOF值低的问题。层状双氢氧化物(LDHs)因其独特的二维结构和丰富的羟基,被视为制备高性能催化剂的有前景的前驱体。
北京化工大学化学学院李殿卿课题组通过剥离块体CoAl-LDHs制备了超薄且富含空位的纳米片,然后将其与氧化石墨烯(graphite oxide, GO)组装,得到了一种杂化催化剂CoAl-ELDH/GO。
文章中作者使用了XAFS一系列表征证明了CoAl-ELDH/GO中存在富氧空位的存在。
图文分析
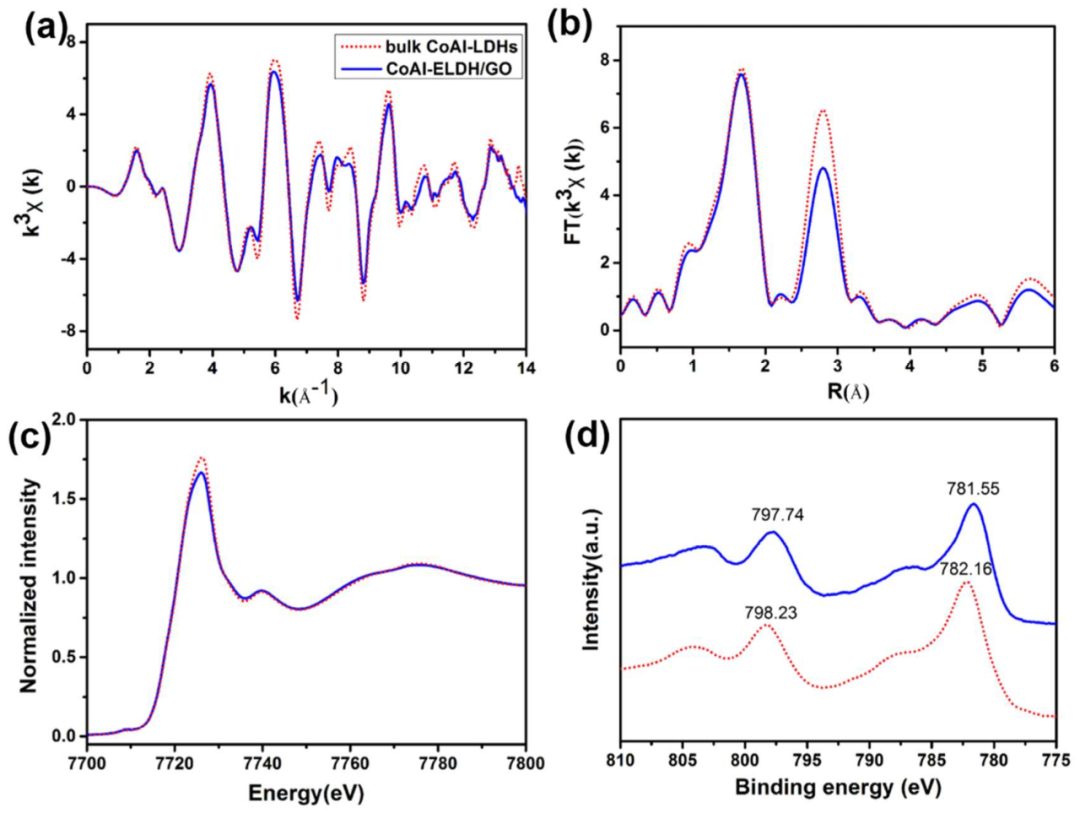
图5(a)CoAlELDH/GO和块体CoAlLDHs的Co K边EXAFS k³χ(k)振荡函数以及(b)相应的傅里叶变换曲线。
X射线吸收精细结构光谱(XAFS)被用来探索材料中的局部原子排列。如图9a所示,CoAlELDH/GO的Co K边扩展X射线吸收精细结构光谱(EXAFS)k³χ(k)振荡曲线与块体LDH非常相似,表明宿主层的ab平面结构得到了很好的保持,这可能源于GO对LDH晶格的保护。
FT k³χ(k)谱图显示1.6 Å和2.8 Å处的两个主要峰,分别对应于最近的Co-OOH和次近的Co-Co配位。CoAlELDH/GO中Co-Co配位的峰强度明显低于块体CoAlLDHs,Co-OOH配位的峰强度略有下降。
对CoAlLDHs数据的拟合得到Co-OOH的平均距离为2.09 Å,配位数为6.0,Co-Co的距离为3.12 Å,配位数为3.8。
对于CoAlELDH/GO,尽管Co-OOH和Co-Co的距离基本保持不变,但Co-OOH和Co-Co的配位数显著下降至5.5和2.8,这表明超薄的纳米片性质导致了新生成的钴和氧空位以及更扭曲的结构,从而形成了配位不饱和的CoO6-x八面体。
从X射线吸收近边结构(XANES)光谱(图9c)可以看出块体LDHs和CoAlELDH/GO中Co中心的表面原子结构的差异。
尽管LDH和ELDH/GO的归一化Co K边XANES光谱在约7726 eV处显示出相同的光子能量峰,但CoAlELDH/GO的白线强度较低,证实了Co原子的电子密度增强。与XPS描述一致。
GO与块体CoAlLDHs显示出相似的湮灭寿命和相对强度。所有三个样品的I3值相当,表明材料中存在的大量缺陷非常相似。然而,CoAlELDH/GO的τ₁(0.1947 ns)比块体CoAlLDHs(0.1829 ns)长,这表明CoAlELDH/GO中存在一种新的小缺陷;这与EXAFS结果一致。
较长的τ₁和增加的I₁值(CoAlELDH/GO为43.0%,块体CoAlLDHs为36.0%)可归因于正电子在钴空位中的湮灭。
原文链接:Ultrathin and Vacancy-rich CoAl-Layered Double Hydroxide/ Graphite Oxide Catalysts: Promotional Effect of Cobalt Vacancies and Oxygen Vacancies in Alcohol Oxidation
https://pubs.acs.org/doi/10.1021/acscatal.7b03655
DOI: 10.1021/acscatal.7b03655
案例三:JACS||原位XAFS识别富氧空位Co3O4在氧进化反应中的动态行为

背景简介
电化学水分解中的氧析出反应(OER)是一个关键但动力学缓慢的步骤,限制了整个水分解的效率。
为了提高OER效率,过渡金属氧化物(TMO)及其衍生物作为有前途的催化剂被广泛研究,尤其是尖晶石结构的Co3O4,因其钴的混合价态而备受关注。
尽管纯Co3O4的本征活性不高,但通过缺陷工程,如引入氧空位(VO),可以显著提升其OER活性。然而,缺陷在催化剂中的具体作用及其在电催化过程中的动态行为尚未完全明确。
近年来,原位/准原位技术的发展为实时监测电催化过程中的结构和功能变化提供了可能,这些技术的应用有助于揭示催化剂表面结构的动态演变和活性中心/中间体的转化,从而深入理解缺陷位点的真实作用和动态行为。
湖南大学王双印课题组通过原位XAFS识别富氧空位Co3O4在氧进化反应中的动态行为。
图文解析
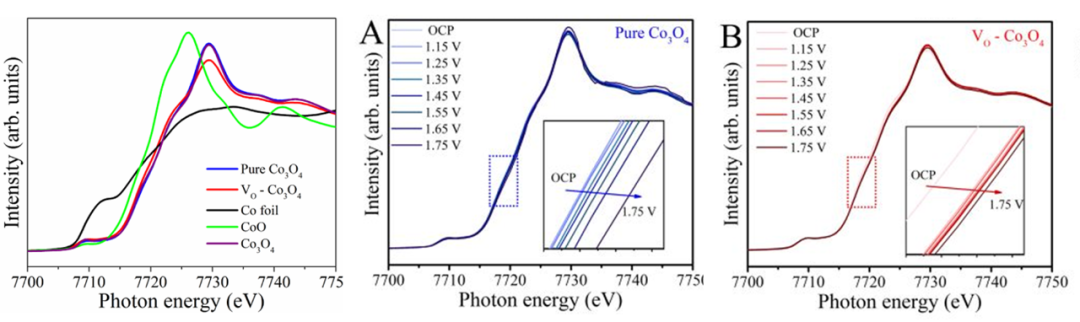
图6 纯 Co3O4(A)和 VO- Co3O4(B)的 Co K 边原位XANES与XAFS谱图。插图分别显示了点状框区域的详细视图。
钴基样品在钴K边记录的X射线吸收近边结构(XANES)光谱所示。VO-Co₃O₄的钴K边能量证实了由于VO的存在,VO– Co3O4中钴的氧化态较低。
为了理解在OER过程中,表面钴位点的潜在依赖结构和氧化态的重建,以及动态缺陷电催化机制,作者在OER过程中进行了原位XAFS实验。在临界电位(从开路电位记录至相对于可逆氢电极1.75V)下收集了钴K边数据,如图10A和B所示。
众所周知,XAFS可用于通过钴K边的前边特征或主共振来评估钴的氧化态,通过主吸收边的一阶导数,利用主共振的上升吸收边来确定钴的价态变化。然而,不同的能量位移趋势表明钴的价态增加不同(见图10A和B)。
钴价态的增加可以被视为催化剂重建表面上的去质子化过程。在氧气逸出之前,与纯Co3O4中钴离子价态的缓慢上升相比,VO– Co3O4中钴离子的价态氧化过程更快。
当阳极电位继续增加,表面电荷无法将Co(IV)氧化到更高的价态时,电子从表面氧原子被移除,导致氧气逸出并伴随结构重建。根据先前文献,由于在高阳极电位下钴位点对O2p电子的吸引力更强,介质中的OH⁻(M-OH*)的去质子化反应变得相对容易。
因此,由于暴露的钴位点的电正性增强,VO很可能促进OH⁻离子在活性钴位点上的吸附,形成吸附的MOH*物种,并随后在低电位(即1.45V)下促进去质子化过程,形成表面活性氧物种(Co-OOH*),这解释了OER活性的增强。
原文链接:Operando Identification of the Dynamic Behavior of Oxygen Vacancy rich Co3O4 for Oxygen Evolution Reaction
https://pubs.acs.org/doi/10.1021/jacs.0c00257
DOI:10.1021/jacs.0c00257