


说明:电催化材料的催化活性高度依赖于过渡金属的d轨道电子构型,特别是eg与t2g轨道的填充情况。在典型的八面体晶体场中,这两类轨道具有不同的能级和反应性。
研究发现,eg轨道电子占据数接近1时,有利于优化中间体的吸附强度,从而提升催化效率。通过密度泛函理论(DFT)计算,可以精确提取eg/t2g的态密度、d带中心及电荷分布,用于构建量化的活性描述符。
顶刊文献中已广泛应用这些电子结构指标分析OER和ORR性能趋势,推动形成一套以轨道调控为核心的材料设计策略,显著提升了电催化剂的可预测性与定向开发能力。
什么是eg与t2g?
过渡金属离子处于配位场时,其价层d轨道会因配体的静电场作用而发生简并裂分,即晶体场效应。以八面体配位为例,金属离子被6个配体围绕,五个本来简并的d轨道将分成两组:能量较低的三个t2g轨道(对应dxy、dyz、dzx,指向配体间隙)和能量较高的两个eg轨道(对应dx2-y2、dz2,指向配体方向)。
这一八面体晶体场裂分能量通常记为10Dq,其大小受金属离子及配体场强度影响。若裂分能量较大,d电子优先成对填充低能的t2g轨道(低自旋态);若裂分能量较小,则电子遵循洪特规则占据高能eg轨道(高自旋态);裂分能适中时可能出现中自旋态。
因此,eg和t2g轨道是晶体场理论下定义的两类轨道组态,反映了过渡金属离子在特定配位对称下d电子的分布情况,它们与配合物的光谱、磁性和稳定性密切相关。
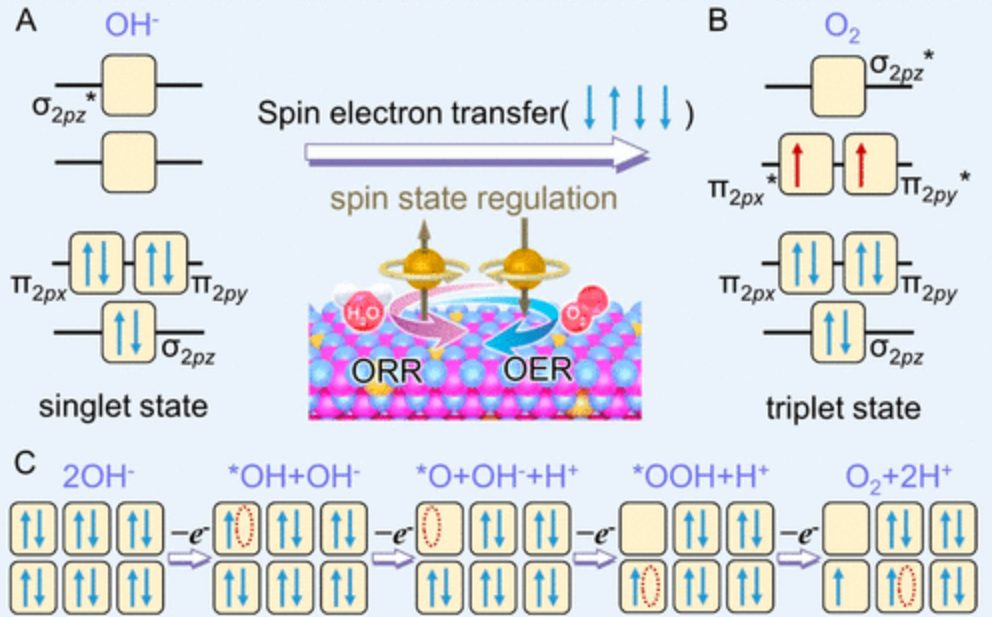
DOI:10.1021/prechem.3c00059
在电催化材料(如过渡金属氧化物和含过渡金属的复合物)中,eg和t2g轨道占据情况被证明是影响催化活性的重要电子结构指标。八面体配位的过渡金属氧化物中,表面过渡金属的eg轨道电子占据数与催化活性呈火山曲线关系。
例如,Shao-Horn等人在Science报道中系统考察了超过10种过渡金属氧化物的析氧反应(OER)本征活性,发现OER活性随表面3d轨道中eg对称轨道占据数呈火山形变化,峰值对应eg占据接近1时的组合。
这意味着适中的eg电子占据(约1个电子)可实现最佳的催化键合强度:过高或过低的eg填充都会降低活性,其中eg全空(0)或全满(2)时活性最低,而接近半填充(~1)时催化效率最高。
机理上,这是由于eg轨道属于金属–氧键的反键轨道,其占据程度直接影响金属对氧中间体的键合强度:eg电子过少会使反键轨道空置,金属与氧键过强,产物难以脱附;而eg电子过多会削弱金属–氧键合,活化吸附物变得困难。
适度的eg填充使金属–氧键强度处于中等范围,符合Sabatier原理,从而在反应能垒和中间体稳定性间达到最佳平衡。
这一电子结构调控思路已被广泛用于解释和优化电催化性能:例如在钙钛矿氧化物中通过调节B位过渡金属的氧化态和自旋态来调整eg电子数,从而优化对氧化物中间体的吸附能。
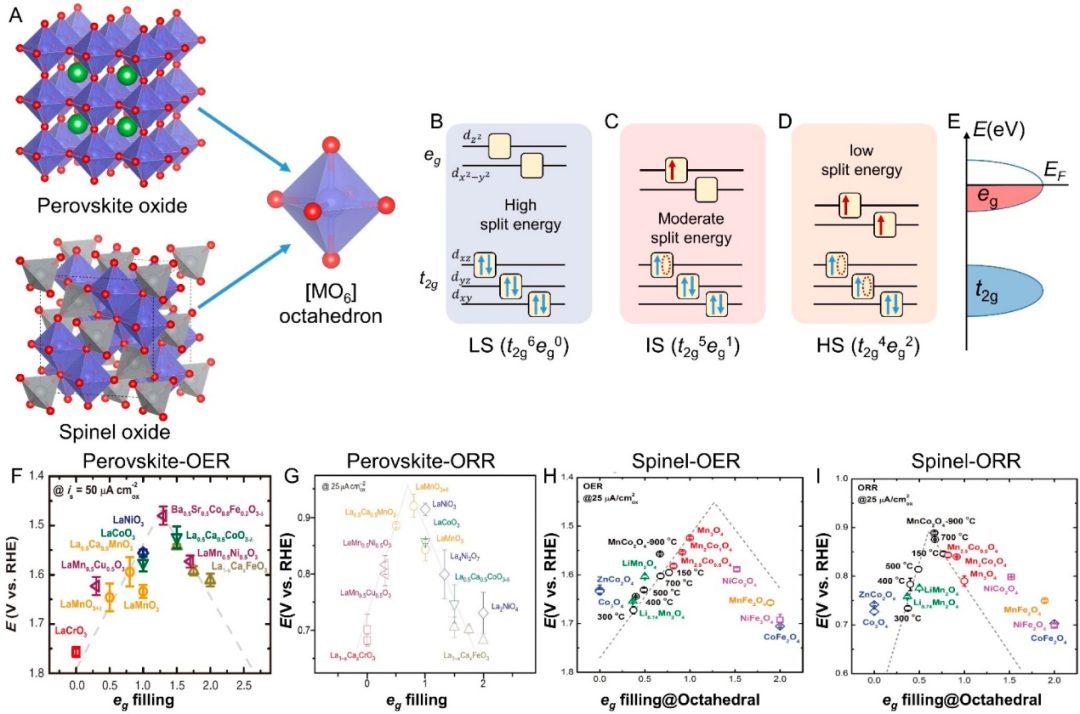
DOI:10.1021/prechem.3c00059
不仅析氧反应,氧还原反应(ORR)的氧化物催化剂也体现出对eg/t2g电子构型的敏感性。研究指出,过渡金属氧化物的ORR活性主要与σ轨道(即eg轨道)的占据数相关,同时过渡金属–氧的共价性作为次要描述符发挥作用。
较高的金属–氧共价性往往伴随适中的eg填充,有利于在ORR机理中平衡表面过氧桥键的生成/解离和氢氧根的再生步骤。
因此,在电催化材料设计中,研究者常以调整eg、t2g轨道电子占据来调控对反应中间体的吸附强度和反应能垒。
例如,通过异价元素掺杂改变过渡金属的d电子数或晶场环境,可以调控活性位的eg/t2g填充,从而影响催化剂对键合物种的电子供给或抽取能力,实现对反应活性的调节。
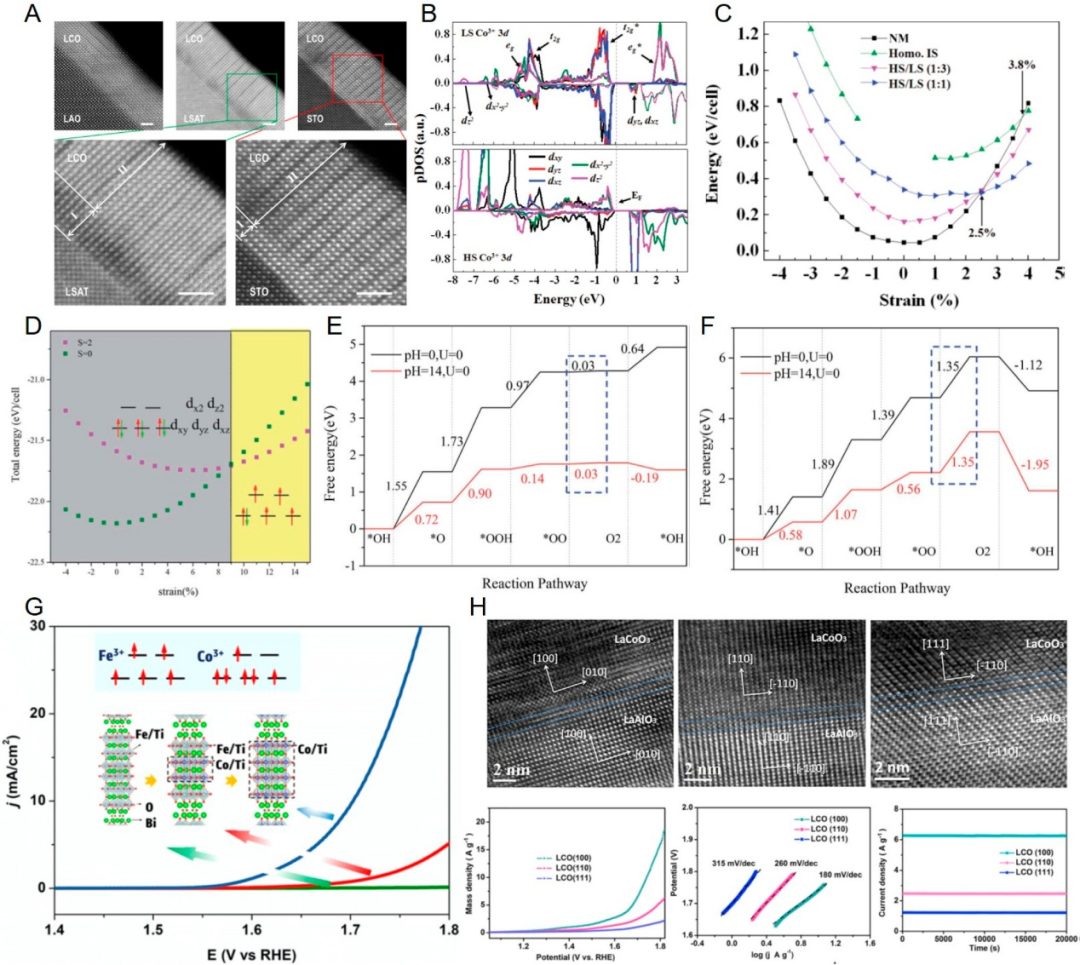
DOI:10.1021/prechem.3c00059
DFT计算中eg/t2g
为了定量研究电催化材料中eg/t2g轨道占据与催化性能的关系,常借助密度泛函理论(DFT)计算提取相关电子结构信息。
将过渡金属原子的d态根据轨道对称性投影到t2g和eg子空间,计算每一组轨道的态密度分布及费米能级以下的积分占据数。
这种方法可以直接得到eg填充程度和t2g态贡献。例如,有研究通过调整ABO钙钛矿的组成,计算不同组成下B位金属d轨道的PDOS,提取出eg轨道电子占据数的变化,并将其作为描述符与催化活性建立关联。
将过渡金属的整体d态密度相对于费米能级计算加权平均能量。d带中心是金属催化表面常用的电子结构描述参数;在过渡金属氧化物中,d带中心结合eg填充可以一起理解金属–氧键强弱。当d带中心接近费米能级且eg有适度占据时,一般意味着与吸附物的键合适中,有利于催化反应。
针对金属氧化物体系,除了金属d带,氧的p带位置同样重要。O 2p中心可作为金属–氧共价性的量化指标:O 2p能级越接近费米能级,表明金属–氧键共价性越强,过渡金属氧化物中孔穴浓度更高。这一指标常与eg占据结合讨论,以解释如高eg填充伴随高共价性对提高OER/ORR活性的作用。
通过DFT计算的电荷分布,将电子归属于各个原子,得到活性过渡金属位的净电荷。Bader电荷可以反映金属的氧化态和电荷转移程度。例如,随掺杂或价态变化,活性中心Bader电荷的变化往往对应d电子占据的增减,从而关联到eg/t2g填充的改变。
虽然Bader电荷不直接区分轨道类型,但结合PDOS分析可用于佐证eg/t2g电子重分布对催化活性的影响。
选取与反应相关的能量窗口,积分该区间内过渡金属d态的局部密度,得到一个定量数值,用于与反应动力学参数相关联。这一方法实质上捕捉了在工作条件下可参与反应的d电子态数量。
例如,JACS研究指出LaMn系列中,Ni/Mn的d态在ORR/OER电位范围内的集成态密度与相应催化反应速率呈线性关系,体现出该积分态密度作为eg/t2g相关指标的有效性。
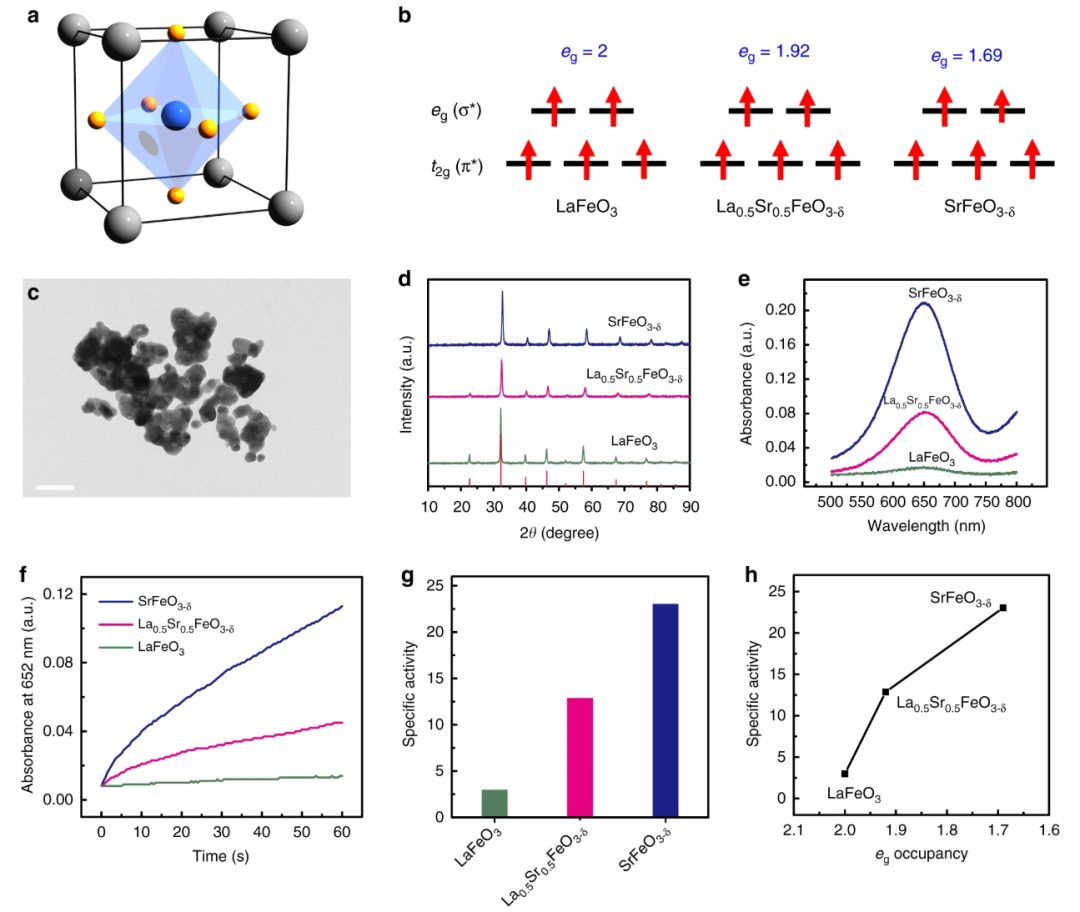
DOI: 10.1038/s41467-019-08657-5
上述方法相结合,可以从DFT计算结果中提取丰富的信息,用于构建电子结构描述符。通过比较不同材料的这些描述符数值,研究者能定量评估eg填充程度、t2g轨道贡献及金属–氧键特性如何影响吸附能和反应能垒,从而指导材料改性和性能优化。
基于eg/t2g活性趋势分析
大量顶级期刊文献(如Nature、Science、JACS、Nature Catalysis等)已经证明了eg/t2g轨道分析在揭示电催化活性趋势中的价值。一系列研究以eg轨道占据数作为核心描述符,成功解释了不同材料催化性能的差异,并用于预测新催化剂。
DOI:10.1039/d4sc05539j
例如,Suntivich 等人在2011年分别发表于Science和Nature Chemistry的开创性工作,提出了以过渡金属表面eg轨道电子占据数为主要判据的设计原则:对于OER,eg占据接近1时表现出峰值活性;对于ORR,eg占据和B位金属–氧共价性的综合考虑能够预测催化剂取代铂的潜力。
这些研究加速了人们在钙钛矿、尖晶石等非贵金属氧化物中寻找高效氧电极催化剂的步伐。此后,不同体系的电催化研究者纷纷引用和拓展这一思路,将d电子构型与催化性能建立定量联系。
在钙钛矿氧化物之外,衍生的新指标也不断涌现。例如,在尖晶石氧化物中,八面体与四面体位点的角色有所不同。
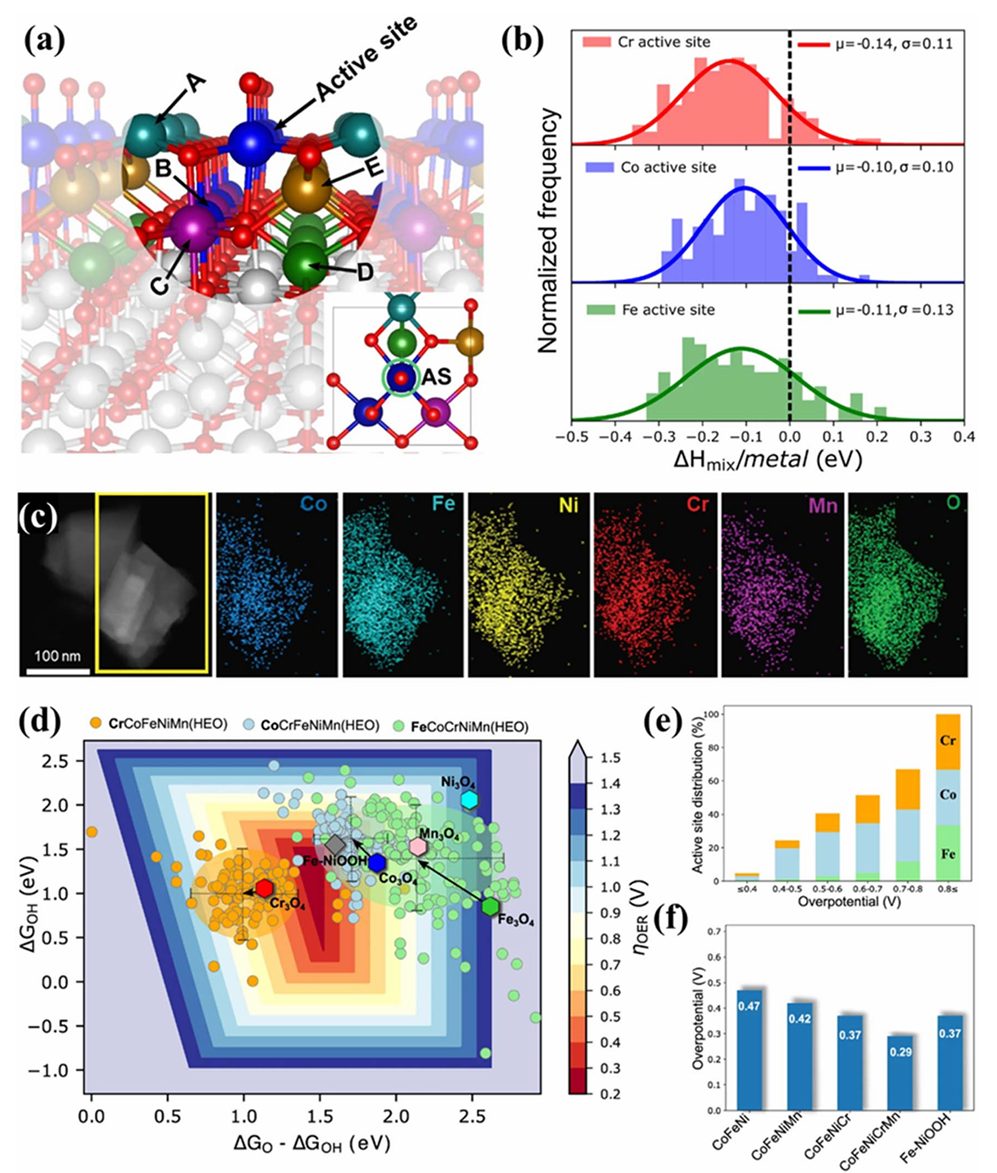
有研究首次提出以“未成对的t2g电子占据数”作为四面体位过渡金属ORR活性的描述符,发现在高熵尖晶石中通过组分熵工程可有效调控该t2g和传统eg描述符,从而同步提升ORR和OER性能。
具体而言,高熵致使Co的t2g电子和Ni的eg–O$2p$轨道发生重新分布,形成协同的双活性位,大幅提高了氧还原反应动力学。
这表明在复杂多金属体系中,t2g轨道的信息也不可忽视,特别是当反应活性位处于不同配位环境(如尖晶石的四面体位)时,未成对电子数可直接影响氧分子吸附与活化过程。
此外,研究者也在探索结合eg/t2g概念的新型综合描述参数。例如,Li等人在近期工作中构建了一个“阳离子/阴离子复杂度指数”,综合考虑了过渡金属与阴离子间的相互作用复杂程度,用于预测金属磷三硫化物催化剂的OER活性。
这一指数实质上反映了材料中金属-d与阴离子p轨道杂化和电荷分布的统计特征,是对传统单一轨道占据描述符的拓展。再比如,有文献采用金属d轨道未占据态密度、活化能等组合参数作为描述符,以弥补d带中心等在强离子性氧化物体系中的不足。
这些新策略体现出电子结构描述的不断演进:从早期关注单一轨道的占据,到兼顾金属–氧协同效应和多轨道参与,从而更全面地捕捉催化活性的本质决定因素。
DOI:10.1039/d4sc05539j
总的来说,当今顶刊关于电催化剂的研究普遍将eg/t2g轨道分析作为重要切入点,无论是直接以eg填充作为火山曲线描述符,还是结合共价性、能带中心等发展综合指标,目的都是为了将材料的电子结构特征与反应动力学之间建立起清晰的关联。
这种基于轨道的分析方法已在过渡金属氧化物、电催化单原子、金属氮化物/硫化物等体系中得到验证,并不断催生新的理论框架和设计原则,以指导高效电催化材料的开发。
eg/t2g与离子比例
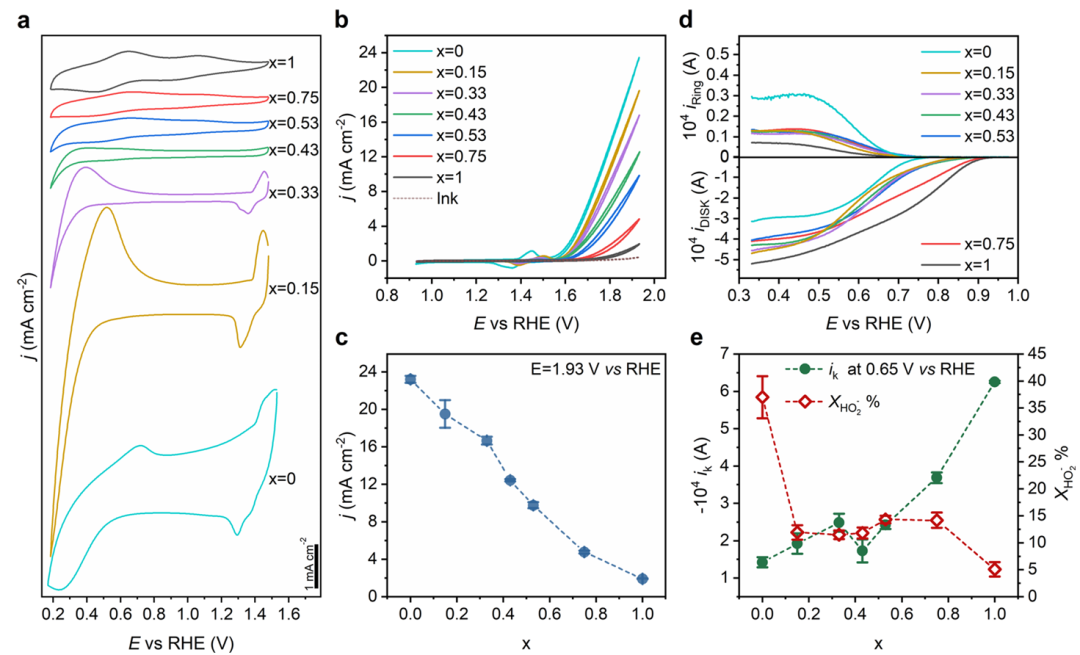
Alkhalifah等报道了一系列钙钛矿氧化物LaMnxNi1–xO3纳米结构,用于ORR/OER双功能催化,并重点解析了材料活性随eg/t2g电子结构的变化趋势。
通过控制B位上Mn/Ni比例,他们调节了过渡金属的d电子占据:Ni3+和 Mn3+离子的比例变化会引起eg轨道占据数和金属–氧共价程度的系统性改变。研究者综合运用实验电化学和DFT计算,对比了一系列成分的氧电催化性能与电子结构描述符。
DOI:10.1021/jacs.1c11757
OER本征活性随着Ni含量升高呈现近似线性提高,并非简单的火山关系。DFT计算显示,这一线性趋势可用过渡金属d态在反应电位区域的积分态密度(IDOS)来解释:Ni/Mn 3d在费米能附近的DOS积分值随Ni含量增加而增大,且与实验测得的OER动力学常数呈线性相关。
这意味着Ni掺入提高了B位d态在价带顶的占据,从而增强了对关键中间体的电荷转移能力,降低了OER反应能垒。
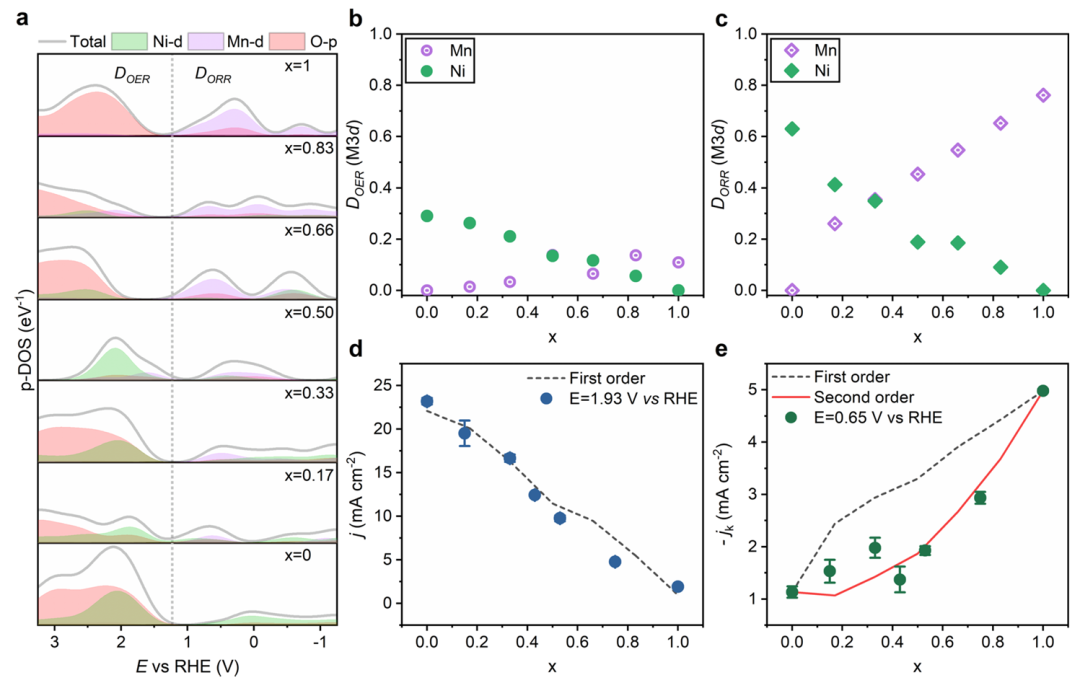
为了深入理解上述关联,作者区分了Ni和Mn各自的d轨道贡献。通过元素分投影DOS,他们发现随Mn替换为Ni,体系的d带中心上移,Ni的eg反键轨道在费米能附近提供了更多未占据态,而Mn的t2g贡献相对减少。
这种轨道组成的变化提高了材料在工作电位下可利用的d电子态数量(即IDOS增大),从而加速了氧-O键的断裂和形成步骤。换言之,Ni的eg轨道参与使表面过渡金属对氧中间体的结合更适宜,提升了催化活性。
有趣的是,该系列材料在ORR中的活性趋势与OER略有不同。作者报告,虽然增加Ni含量有利于OER,但ORR活性在高Ni含量时并未单调提升,而是受到Mn^3+含量的影响。
这被归因于ORR过程中可能涉及过氧物种的稳定化机制,与eg轨道占据和金属–氧键强度的关系更为复杂。因此,作者提出需要综合考虑eg占据和金属–氧共价性来优化双功能催化性能。
DOI:10.1021/jacs.1c11757
综上,这篇论文通过控制组成调整eg/t2g电子占据,并借助DFT定量分析了轨道态密度与反应速率的关系,展示了基于轨道描述符进行催材设计的范例。作者的方法将传统的eg填充概念拓展为更普适的“轨道积分态密度”描述符,在复杂体系中仍能成功预测和解释催化活性变化。
这一案例凸显了电子结构–性能关联研究的威力:通过深入理解eg、t2g等轨道的作用,研究者不仅解释了实验现象,还提出了明确的材料设计准则。这对今后开发高效电催化剂具有重要指导意义,说明顶刊研究中eg/t2g轨道理论已成为电催化领域阐释机理与指导材料创新的关键工具之一。
总结
在电催化研究中,eg与t2g轨道作为晶体场理论下的关键电子构型,正日益成为解释和预测催化活性的核心指标。过渡金属离子在八面体配位环境中,其五个d轨道裂分为能量较低的t2g
与能量较高的eg轨道,其中eg轨道因其反键特性直接参与与反应中间体的电子相互作用。
研究发现,催化性能常随eg轨道电子占据数呈火山型变化,最佳活性对应eg占据接近1,这一规律被广泛用于OER和ORR催化剂的筛选和设计中。
近年来,DFT计算在电子结构定量化方面提供了强有力的工具,常通过PDOS轨道投影、d带中心、Bader电荷及积分态密度等方法,解析eg/t2g填充对吸附能、反应能垒及金属–氧共价性的影响。
顶刊文献等系统性研究表明,eg与t2g轨道占据不仅可作为独立描述符,也常与O 2p带中心、杂化强度等组合,用于构建更全面的活性预测模型。