

说明:X射线吸收光谱(XAS)能够确定物质的局部原子结构和电子结构。本文通过同步辐射XAFS技术(含XANES与EXAFS)结合HAADF-STEM、XRD等表征手段,揭示了PtCu/MgO催化剂在CO氧化中的动态结构演变。更多有关知识请看以往推文:
什么是同步辐射R空间?
精选干货|同步辐射PDF基础知识及经典应用分析!
X射线吸收光谱(XAS)测量物质在X射线区域的透射率作为入射光子能量E的函数。它具有元素选择性和轨道选择性,能够确定物质的局部原子结构和电子结构。
XAS可以推导出氧化态、态密度、配位、键长、热参数以及局部环境中的无序性,其精度在原则上比其他技术更高。
由于XAS不依赖于长程有序性,因此特别适合研究具有局部短程有序性的无序系统,例如玻璃、溶液、液体和非晶态固体。
它也可以应用于定义明确的甚至理想的晶体。XAS可以确认其他技术所提出的结构,或者推翻一个假设,以支持基于吸收原子物种周围的纳米结构、局部键合和纳米环境的有效局部结构。
尽管XAS可以使用实验室X射线源进行,但它更常在同步辐射光源处进行。大多数同步辐射光源都有专门用于XAS的光束线,XAS是同步辐射的三种最常见技术之一,与粉末衍射和单晶衍射并列。它与其他许多技术互补,能够提供独特的见解。
XAS(X射线吸收光谱)包括X射线吸收精细结构(XAFS),整个XAS光谱可以分为两个区域(图1d):X射线吸收近边结构(XANES)和扩展XAFS(EXAFS)。
XANES通常通过指纹法或线性成分拟合进行研究,涉及复杂的激发理论,使用分子轨道原理或光电子的多路径、多腿散射(双腿或三腿路径,多次散射)。EXAFS区域则可以用现有的多种理论和软件包进行拟合。
在XAS中观察到的复杂振荡结构源于从光吸收边上方发射的光电子波(图1a)与来自邻近电荷密度的弹性散射光电子波(图1c)之间的量子干涉。这可以确定吸收原子周围局部环境的氧化态、几何结构和结构。
此外,XANES区域还包括前边特征,这些特征对应于束缚态之间的共振激发,可以解释吸收原子的几何结构、配位和氧化态。XAS光谱也可以通过荧光检测(图1b)、电子产额和散射过程间接获得,而无需直接测量X射线吸收。
一些实验设置使用部分荧光产额或荧光检测,并选择特定的发射线对应的区域作为感兴趣区域。
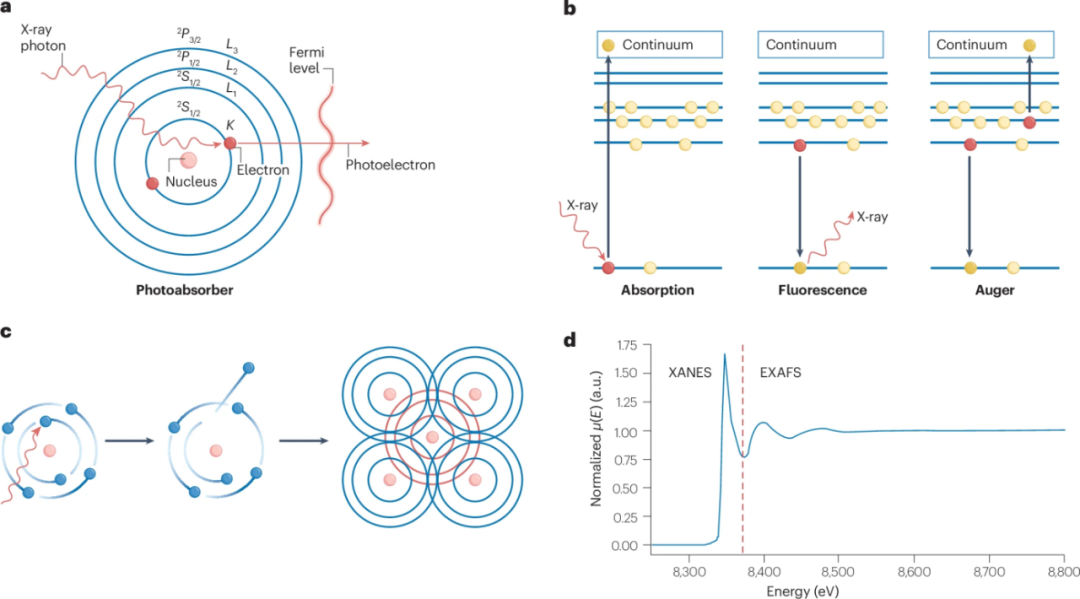

图1 X射线吸收光谱学中的过程
XAFS光谱学研究了由于化学键合而引起的X射线吸收系数的变化。通过选择具有特征吸收边能量的原子,可以针对不同的原子元素进行研究。
XAFS的一个显著优势在于,它不需要样品具有长程有序性,这与传统的衍射方法形成对比。因此,XAFS适用于晶体、非晶态系统、多晶固体、合金、溶液和分子气体等多种物质状态。
此外,XAFS对样品或实验环境的要求并不苛刻,这使得它能够用于研究非理想的真实世界体系。
可靠的XAFS测量必须考虑X射线的一般行为及其与物质的相互作用——吸收和散射的能量依赖性——以及XAFS测量过程的特定特性,例如记录光谱所需的能量范围、采样和分辨率。
透射模式是XAFS测量中最简单且最常用的方式,其原理类似于基本的分光光度测量。然而,与实验室规模的仪器不同,同步辐射X射线源可能有数百米的直径,而仪器的光束线则有数十米的长度。
样品的尺寸通常在1 cm左右,但通过合适的聚焦仪器,X射线束可以达到小于1 μm的尺寸。
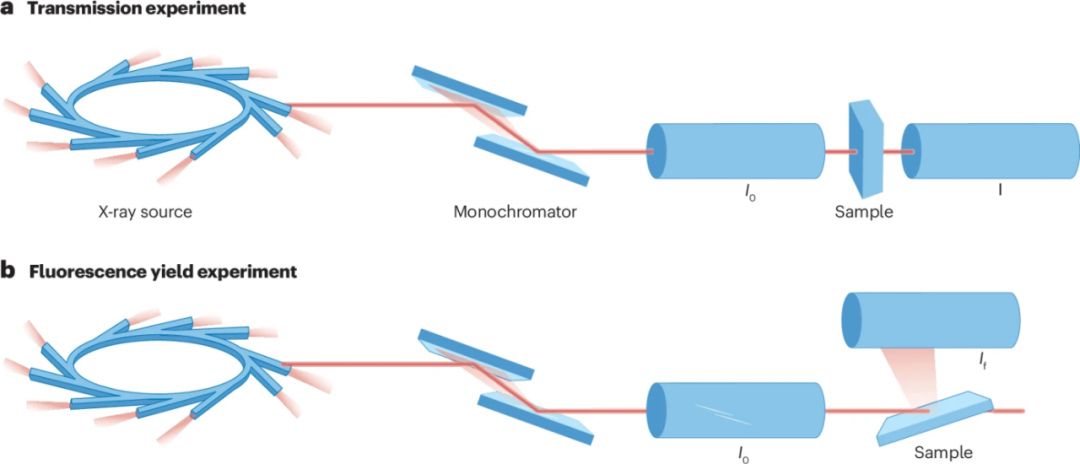
图2 X射线吸收精细结构测量的示意图
XANES区域覆盖了吸收光谱中较低能量的部分,从吸收边以下开始,一直延伸到吸收边以上大约30 eV的位置(图1)。
在这个区域内,多重散射过程具有重要意义。所谓多重散射,指的是被激发的光电子在返回吸收原子之前,会依次被多个相邻原子散射。在这个过程中,光电子会从一个原子(单次散射,双路径)散射到另一个原子(双次散射,三路径),然后再返回到最初发生光吸收的位置,这是因为非弹性损失相对较弱。
XANES区域包括上升边,其中既包含了电离的阈值,也包含了更高能量的成键态,以及前边特征,这些前边特征对应于跃迁到束缚的未填充轨道。
XANES区域包含了关于吸收原子的电子结构(氧化态和态密度)、配位对称性(四面体和八面体)以及轨道占据的信息。
EXAFS区域从吸收阈值以上约30 eV处开始,并延伸数百电子伏特,是调制吸收光谱的振荡结构。通常对EXAFS函数χ(k)进行拟合和分析。
一些软件包同时拟合χ(k)、k²χ(k)、k³χ(k)和χ(r),尽管这在理论上存在挑战。通常使用傅里叶变换(FT)将光谱建模和分析到R空间,即χ(r)与r的关系,以显示径向密度函数χ(r)。
通过滤波和反变换,光谱可以转换到Q空间以拟合χ(q)与k的关系。传统的理论代码如FEFF、GNXAS和EXCURVE用于计算理论光谱,然后将其拟合到实验数据。
随着像FDMX这样的软件包的出现,拟合可以在E空间直接进行,即[μ/ρ](E)、[μ/ρ]pe(E)、μ(E)或μpe(E)与E的关系,包括对近边、XANES和前边的拟合。应对每个确定的参数进行统计分析,以估计其置信区间。
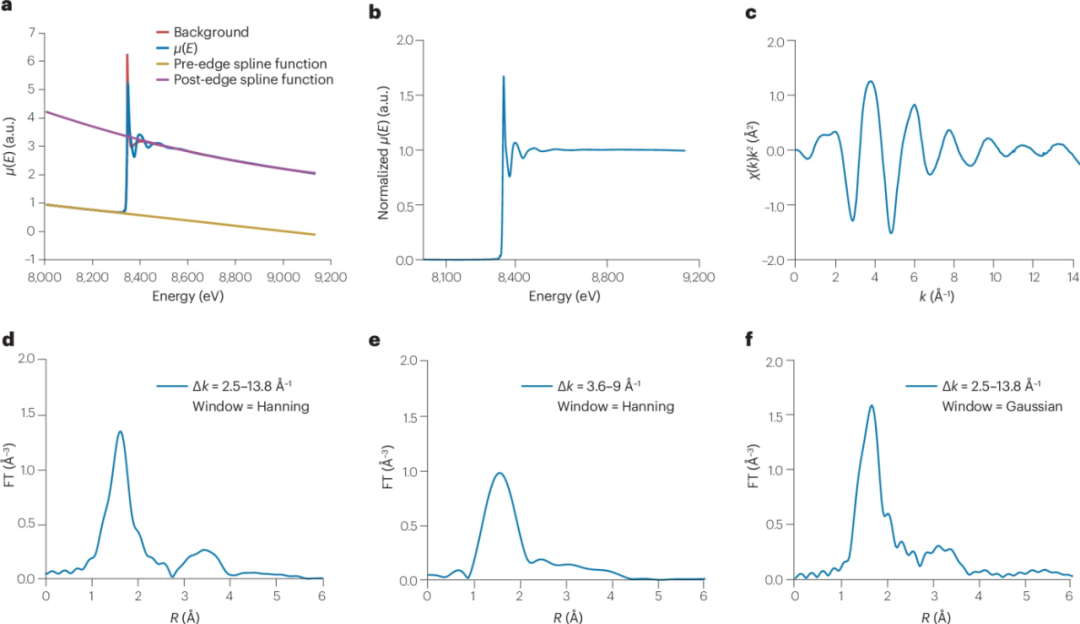
图3 从原始X射线吸收光谱数据中提取和归一化扩展X射线吸收精细结构(EXAFS)光谱的示例
中国科学院上海应用物理研究所李丽娜、南兵和蒋罗震等采用共浸渍法制备了MgO负载的PtCu双金属催化剂,结合各种原位表征技术,揭示了PtCu/MgO催化剂在CO氧化过程中转化为CuOx/PtCu合金(CO氧化的主要活性中心),并且CuOx和PtCu合金在不同温度范围内对CO氧化起着不同的作用。
在低温(条件下,由于CuOx组分含有丰富的活性氧,可以促进CO的氧化;随着反应温度的升高,PtCu合金有效地吸附了CO分子和游离O2气体,促进了CO的转化。(DOI:10.1038/s41467-024-49968-6)
研究人员通过XRD、HAADF-STEM和H2-TPR三种表征手段对PtCu/MgO催化剂进行了分析。XRD结果显示铂和铜高度分散在氧化镁载体上,未形成明显的晶体颗粒。
HAADF-STEM图像表明Pt和Cu在MgO表面均匀分布,形成了平均粒径为1.4 ± 0.3 nm的PtxCuyOz二元氧化物团簇。H₂-TPR检测到187°C的强还原峰,归属于Pt-[O]x-Cu结构,揭示了铂和铜之间存在强相互作用,这种相互作用显著增强了催化剂的还原性。
这些结果表明,PtCu/MgO催化剂具有高度分散的纳米团簇结构和良好的金属间相互作用,为其优异的催化性能提供了结构基础。
XAFS提供了关于铜和铂的精确的电子结构和配位信息
XANES分析显示,Pt/MgO的白线强度高于PtCu/MgO,表明PtCu/MgO中Pt的平均氧化态为3.8,低于Pt/MgO的4。
EXAFS拟合结果表明,PtCu/MgO中存在强烈的Pt-O和Pt-O-Cu配位,证实了PtxCuyOz二元氧化物团簇的形成,且未检测到孤立的PtOx团簇。Cu K边XANES光谱显示Cu/MgO和PtCu/MgO中Cu的平均氧化态均为+2。
由于孤立的CuOₓ物种更为丰富,XAFS分析中Cu-O-Pt配位壳层的拟合较为困难,主要检测到Cu-O和Cu-O-Mg配位结构。
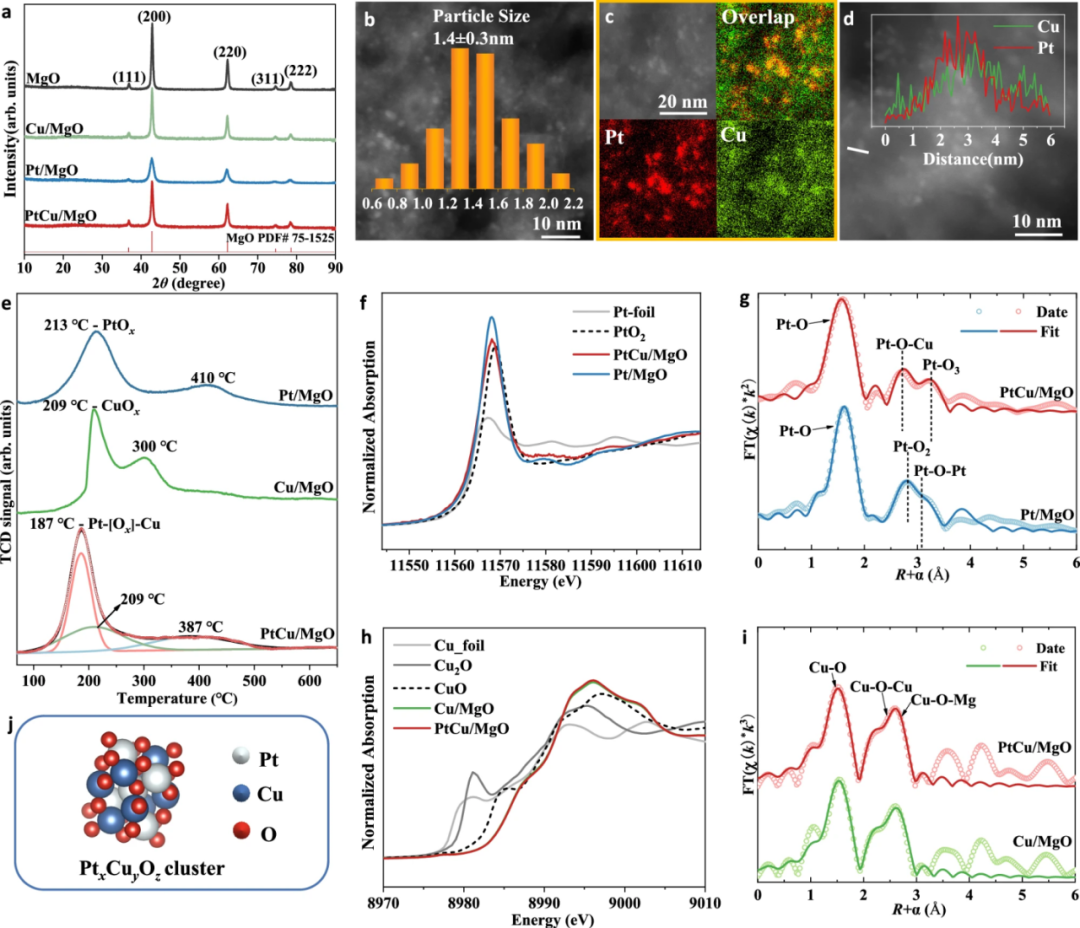
图4 PtCu/MgO催化剂的结构表征
原位XAFS、EDS、XRD结合证实氢气还原后催化剂的结构
氢气还原后,PtCu/MgO催化剂的活性位点尺寸保持在约1.8 nm,且其颗粒为PtCu合金,具有清晰的0.22 nm(111)晶面间距。
EDS元素分布图显示Pt和Cu元素分布一致。XRD图谱中出现的额外峰(40.1°和50.4°)分别对应PtCu合金的(111)晶面和金属Cu的(200)晶面,进一步证实了PtCu合金的形成。
原位XANES结果表明,氢气还原后,PtCu/MgO中Cu和Pt的平均氧化态均为0价。原位EXAFS分析显示,PtCu/MgO(H2)的Pt L3边在约2.60 Å处出现显著峰,配位数约为11.8,与Pt箔明显不同,且与CuPt合金的标准Pt-Cu路径一致;Cu K边EXAFS在约2.59 Å处出现Cu-Cu壳层峰,配位数约为6.3,表明氧化铜团簇被还原为金属Cu颗粒。
小波变换(WT)分析进一步证实了Pt-Cu合金的形成,其WT等高线图在约7 Å⁻¹处的强度最大值归因于Pt-Cu配位。结合HAADF-STEM、准原位同步辐射XRD和原位XAFS表征结果,证实了氢气还原后PtCu/MgO中形成了PtCu合金。
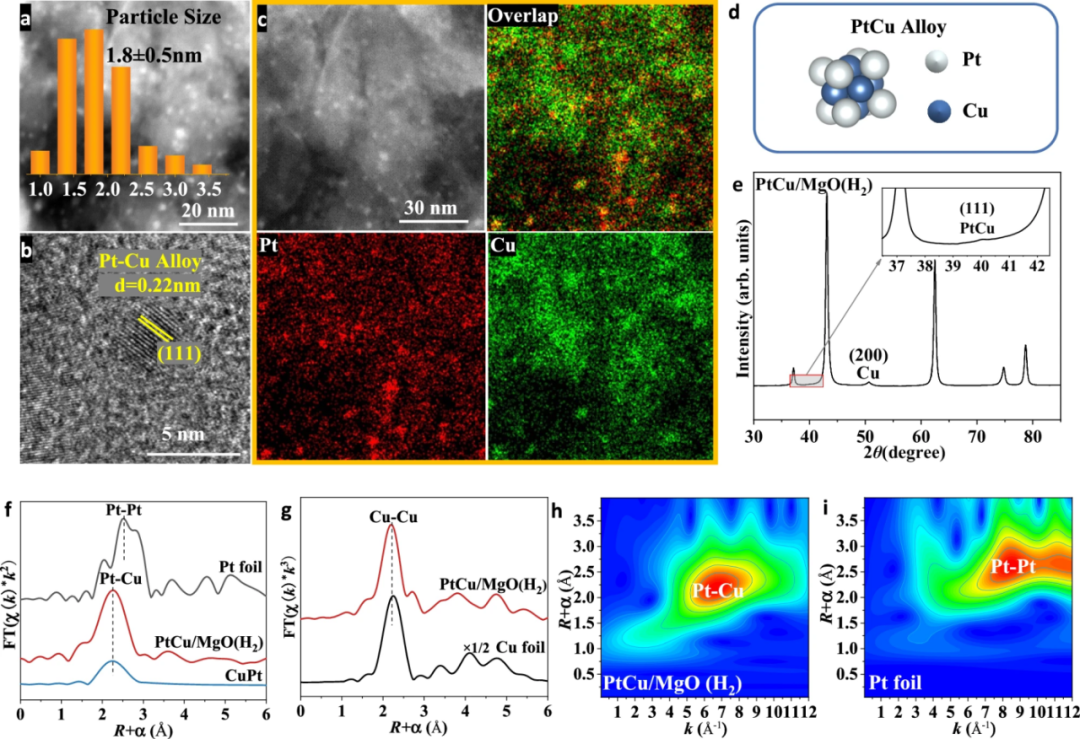
图5 氢气还原后PtCu/MgO的结构表征
HAADF与原位XAFS表征CO氧化过程中PtCu/MgO催化剂活性位点的配位结构与氧化态
CO氧化反应后,经过像差校正的HAADF-STEM图像显示,PtCu/MgO(使用后)的活性位点平均粒径为1.9 ± 0.6 nm,而Pt/MgO(使用后)为1.7 ± 0.4 nm。
HRTEM图像表明PtCu/MgO使用后的活性位点为Pt-Cu合金,其(111)晶面的d间距为0.22 nm,且IFFT图案和线强度分布也证实了这一点。此外,PtCu合金颗粒的线扫描结果表明Pt和Cu在合金中均匀分布,说明PtCu合金结构在CO氧化过程中保持稳定。
准原位同步辐射XRD图谱进一步证实了PtCu合金的存在,其(111)晶面的衍射信号位于40.1°。对于Pt/MgO,主要活性位点为金属铂,其(111)晶面的d间距为0.23 nm。
原位XAFS技术揭示了PtCu/MgO在CO氧化过程中的结构变化。在90°C时,Pt-Cu壳层稳定,未形成Pt-O壳层。随着温度升高,铂物种轻微氧化,氧化态在150°C和270°C时分别为+0.8和+1.8。
XPS也证实了铂物种的氧化。在时,PtCu合金结构稳定。对于Cu物种,500°C氢气还原后还原为金属Cu,但在90°C CO氧化条件下形成CuOx物种。150°C时,Cu物种再氧化并分散。
相比之下,Cu/MgO在90°C CO氧化时仍保持金属态,完全分散为CuOx需更高温度。这表明PtCu合金中的Cu原子在CO氧化过程中能快速解离O2并形成表面CuOx物种。
多次CO氧化循环后,催化剂活性略有下降,可能是由于长时间氧化导致PtCu合金降解,但可通过氢气还原恢复活性。
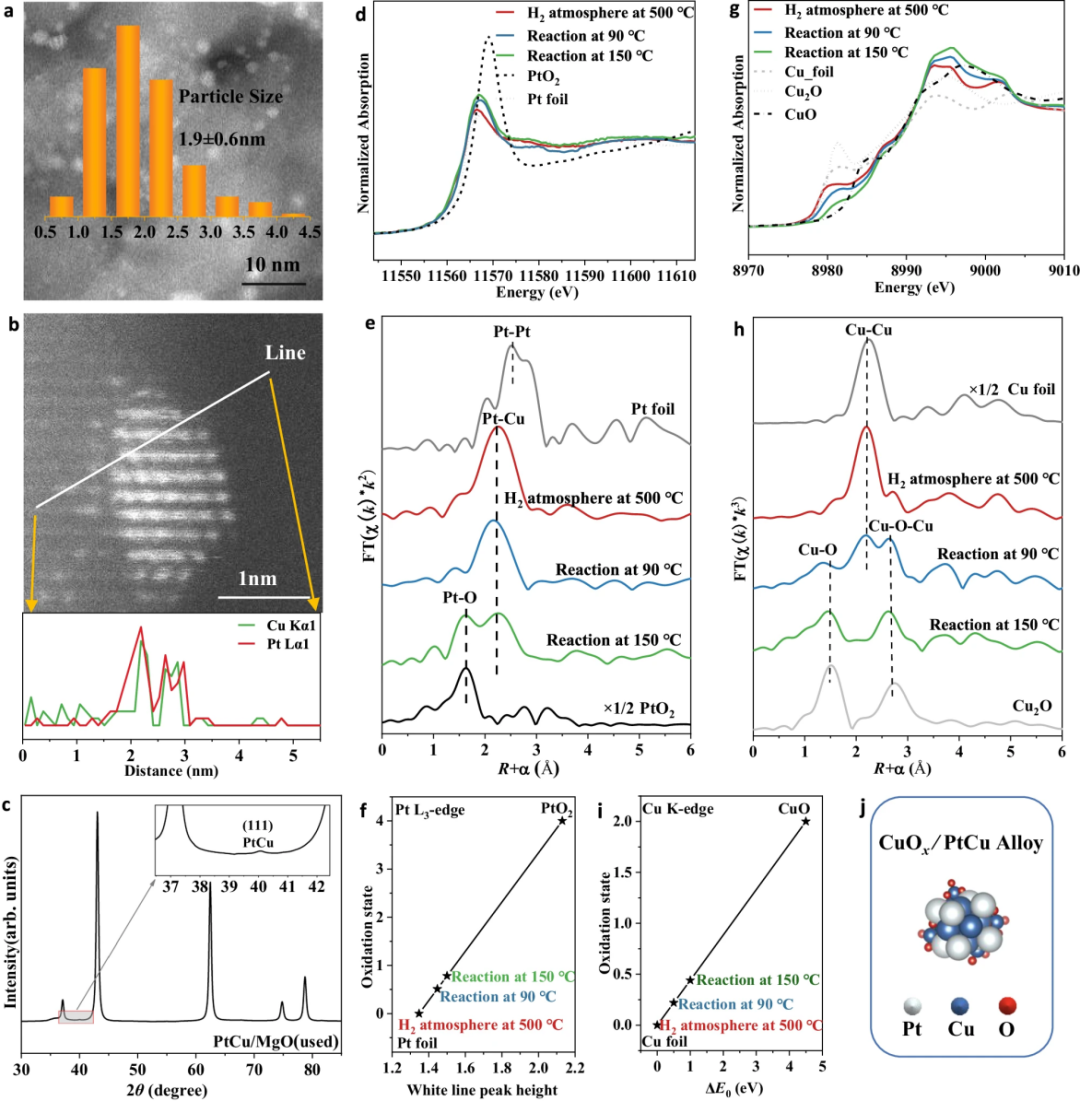
图6 CO氧化过程中PtCu/MgO催化剂的结构表征
从PtCu合金的稳定结构到CO氧化机制的阐明,同步辐射XAFS技术在该研究中展现了其作为催化剂结构表征的强大能力。
它与HAADF-STEM、XRD、XPS等技术的协同应用,成功揭示了PtCu/MgO催化剂在CO氧化过程中的活性位点演变和结构稳定性。
通过精确分析Pt-Cu配位结构和氧化态的变化,不仅证实了PtCu合金在低温下的高效催化性能,还阐明了Cu物种在氧化过程中快速解离O2并形成表面CuOx物种的机制,最终实现了高效、低温的CO氧化转化。
随着同步辐射技术的不断进步,其将在催化反应机理研究、催化剂设计与优化等领域持续发挥关键作用。