

定义与结构
富锂锰基氧化物是一类在电池领域极具研究价值与应用潜力,拥有独特晶体结构和卓越电化学性能的正极材料。在当前全球大力发展新能源产业的大背景下,电池技术作为关键支撑,富锂锰基氧化物的研究进展对提升电池性能、推动新能源产业迈向新高度有着不可忽视的意义。
其通式精准地表示为xLi2MnO3·(1 – x)LiMO2(在这一表达式里,M一般代表的是Mn、Ni、Co等过渡金属元素。
Mn元素凭借其多样的价态变化,能够在充放电过程中灵活地进行电子得失,对材料的容量和循环稳定性有着关键影响;Ni元素因其较高的理论比容量,有助于提升材料整体的能量密度;Co元素则在增强材料的结构稳定性和电子电导率方面发挥着重要作用。
同时限定条件为0,x的取值范围决定了两种结构单元的比例,进而影响材料整体性能 )。这种材料之所以在性能上表现突出,与其复杂的晶体结构密切相关。
它通常是由Li2MnO3和LiMO2两种结构单元巧妙复合而成,Li2MnO3结构单元具有稳定的晶格框架,就像坚固的房屋框架一样,为材料提供了一定的结构稳定性,使得材料在多次充放电循环过程中依然能保持基本的结构形态。
而LiMO2结构单元则在离子传输和电荷转移过程中发挥着重要作用,如同高速公路一般,保障锂离子在材料内部能够快速、高效地移动,以及电荷的顺利转移 ,二者协同合作,共同赋予了富锂锰基氧化物独特的性能优势 。
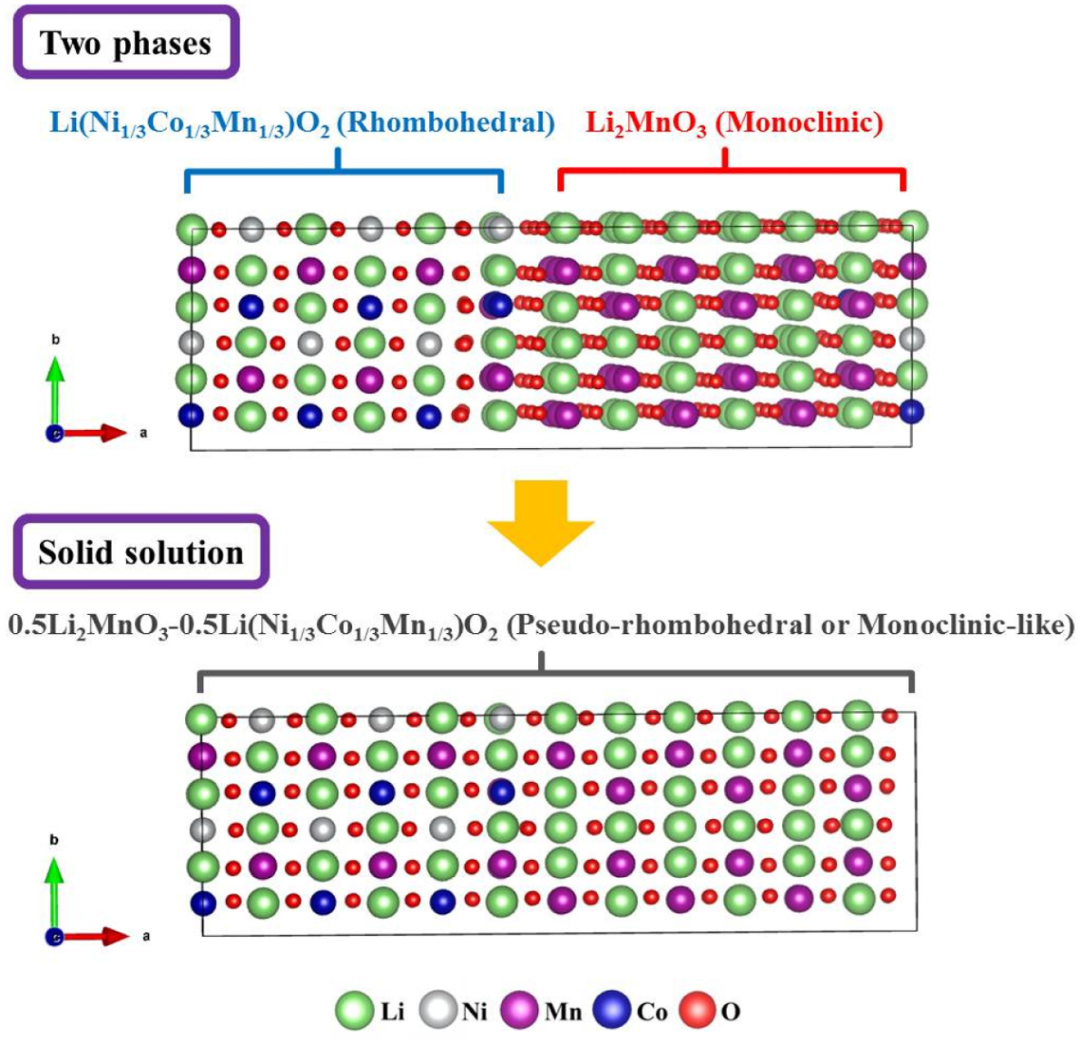

Li2MnO3具有单斜晶系结构(空间群C2/m) ,其结构中锂离子占据了部分过渡金属层的八面体位置,形成了一种特殊的超结构。在这种结构中,氧原子以近似密堆积的方式排列,锂离子和过渡金属离子分布在氧原子构成的八面体和四面体间隙中。
而LiMO2一般具有六方晶系的层状结构(空间群R-3m),锂离子和过渡金属离子交替分布在由氧原子组成的层间,形成了二维的锂离子扩散通道。在富锂锰基氧化物中,这两种结构单元相互交织,使得材料兼具两者的特点,同时也赋予了其独特的电化学性能 。
电子结构特性
通过DFT计算,富锂锰基氧化物的能带结构展现出丰富的信息,为理解其电学性能提供了微观层面的依据。
在典型的富锂锰基氧化物如Li1.2Mn0.54Ni0.13Co0.13O2的能带结构和态密度中,在价带区域,O的2p态密度在较低能量范围内占据主导地位,这表明O原子的2p电子对材料的稳定性起着重要作用。
O的2p轨道与过渡金属的3d轨道在一定能量范围内存在明显的重叠,这体现了它们之间的强杂化作用 。这种杂化作用增强了金属 – 氧键的强度,对材料的结构稳定性和电化学性能产生重要影响。
在过渡金属的3d态密度中,Mn、Ni、Co各自展现出独特的分布特征。Mn的3d态密度在价带中具有多个峰,这与Mn的多种氧化态(Mn3+、Mn4+等)相关。在充放电过程中,Mn的氧化态会发生变化,其3d态密度也会相应改变,从而影响材料的电子结构和反应活性。
Ni和Co的3d态密度同样对材料的性能有重要贡献。Co的存在则有助于稳定材料的结构,并在一定程度上调节材料的电子结构,其3d态密度的分布影响着材料的电子传导和离子扩散性能 。
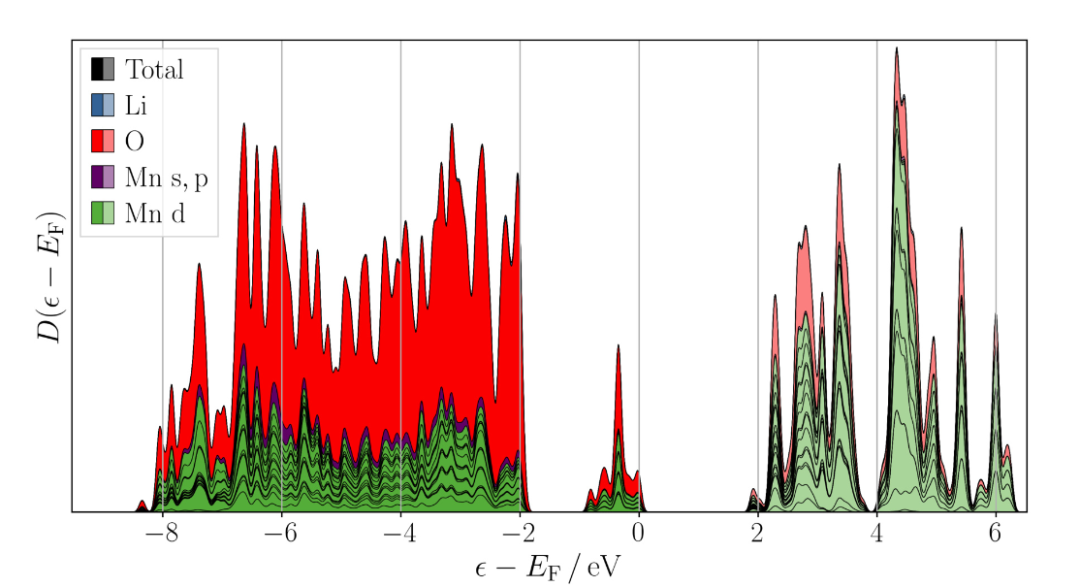
费米能级处的态密度对材料的化学反应活性至关重要。较高的态密度意味着在费米能级附近存在更多可供参与反应的电子,材料具有较高的化学反应活性。在富锂锰基氧化物中,费米能级处的态密度受到多种因素的影响,如过渡金属的种类和含量、晶体结构的缺陷等。
通过调整这些因素,可以有效地调控材料的化学反应活性,例如在材料中引入适量的缺陷或杂质,可以改变费米能级处的态密度,从而提高材料在充放电过程中的反应动力学性能。
Li⁺扩散路径与能垒
利用DFT计算可以精确地确定Li+在富锂锰基氧化物中的扩散路径和扩散能垒,这对于理解电池性能具有重要意义。
在富锂锰基氧化物的晶体结构中,Li+的扩散主要沿着特定的通道进行。以常见的层状结构富锂锰基氧化物为例,Li+通常在由氧原子构成的层间通道中扩散。
通过构建不同的原子模型并进行DFT计算,可以清晰地描绘出Li+的扩散路径。在扩散过程中,Li+需要克服一定的能量障碍,即扩散能垒。扩散能垒的大小直接影响着Li+的扩散速率,进而影响电池的充放电性能。
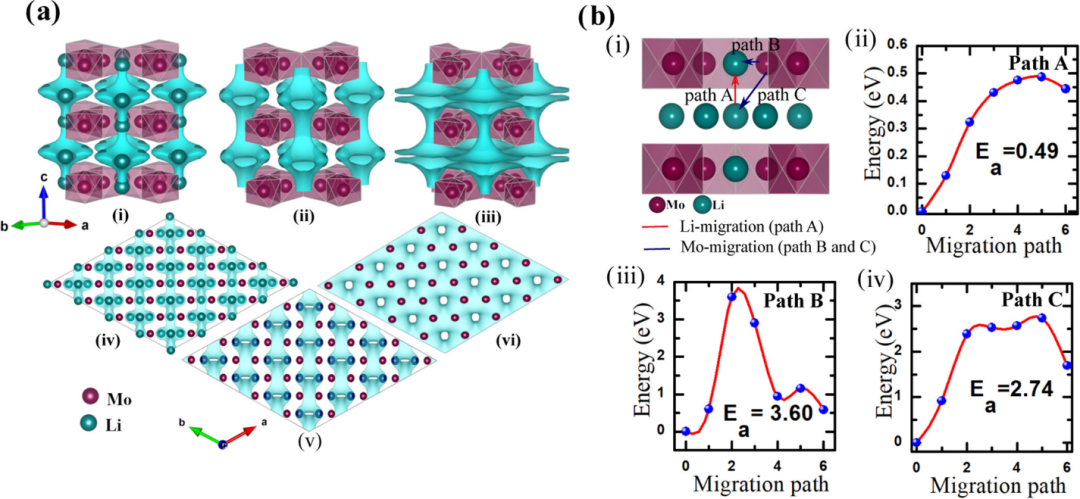
计算结果表明,在理想的晶体结构中,Li+沿着层间通道扩散时,扩散能垒相对较低,有利于Li+的快速迁移。然而,实际材料中存在着各种缺陷和杂质,这些因素会显著改变Li+的扩散路径和能垒。
例如,晶体中的空位缺陷可能会捕获Li+,使得Li+需要额外的能量才能从空位中脱离并继续扩散,从而增加了扩散能垒。
不同晶面方向上的Li+扩散能垒也存在差异。在某些晶面方向上,由于原子排列的特殊性,Li+的扩散能垒可能较高,这会导致Li+在该方向上的扩散速率较慢。
这种各向异性的扩散特性对电池的性能有着重要影响,尤其是在高倍率充放电条件下,Li+扩散的各向异性可能导致电池内部的极化现象加剧,降低电池的充放电效率和循环寿命。
DFT计算揭示调控机制
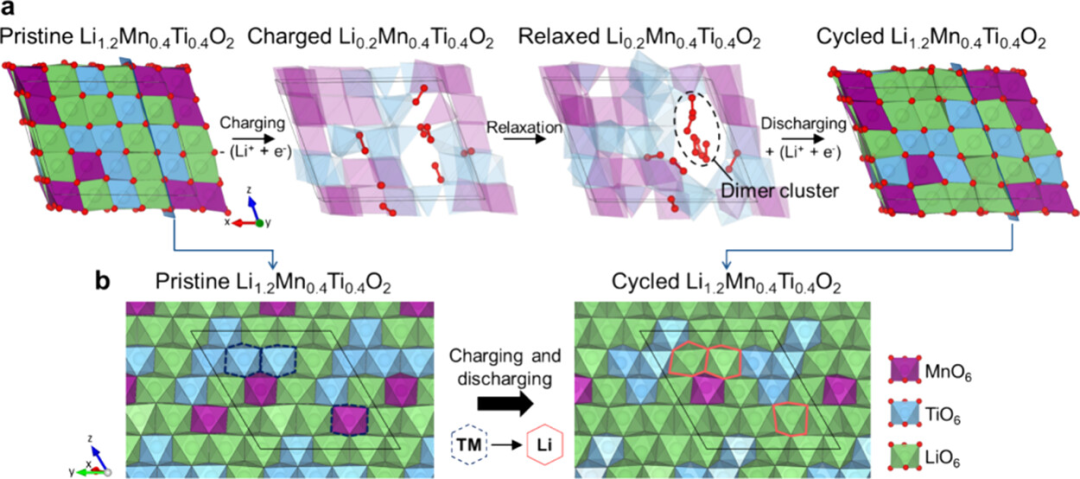
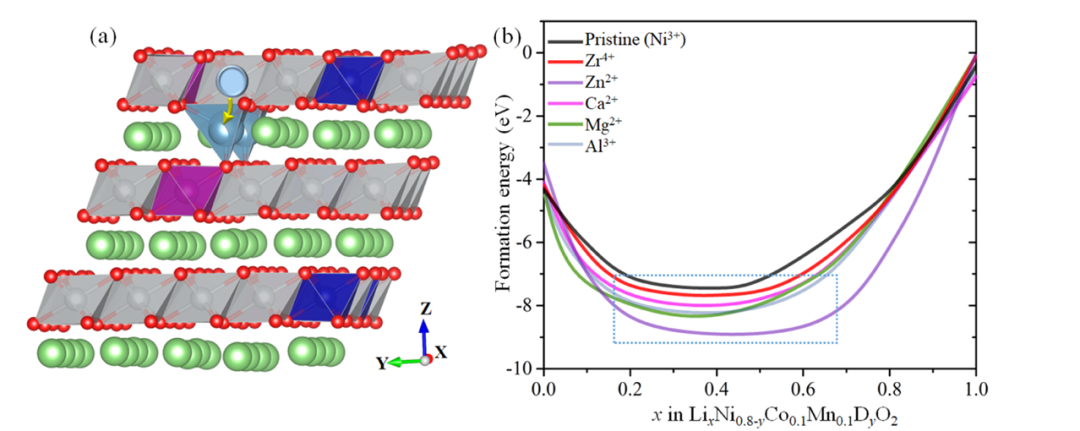
这篇论文通过系统的密度泛函理论(DFT)计算,深入研究了不同阳离子掺杂(Zr⁴⁺、Zn²⁺、Ca²⁺、Mg²⁺、Al³⁺)对富镍层状正极材料LiNi₀.₈Co₀.₁Mn₀.₁O₂(NCM811)结构稳定性和电化学性能的影响。
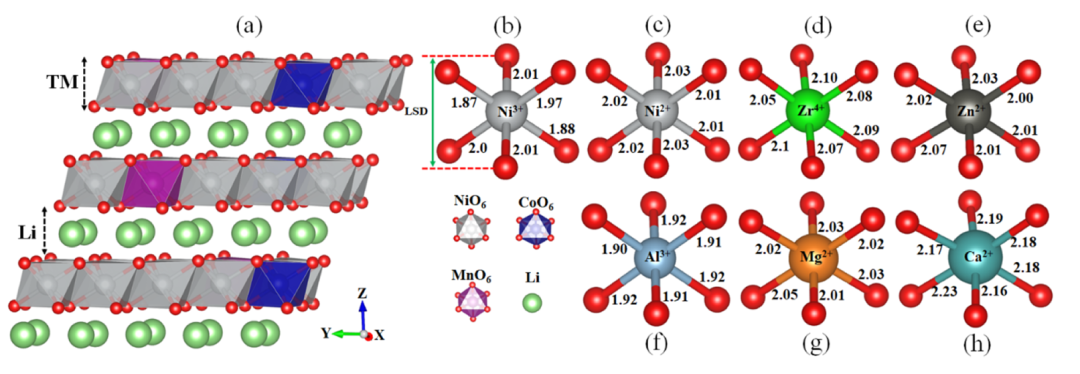
图1展示了优化后的NCM811晶体结构及其键长测量结果,为后续分析奠定了结构基础。通过比较Ni³⁺–O、Ni²⁺–O以及各掺杂元素与O的键长,发现Al³⁺–O键较短(1.90–1.92 Å),而Ca²⁺–O键较长(2.16–2.23 Å),这种键长差异直接影响了Li层的局部间距和整体结构稳定性。
例如,Al³⁺的强键合特性会导致Li层收缩,而Ca²⁺和Zr⁴⁺的较长键长则有助于维持或扩大Li层间距,从而为Li⁺扩散提供更多空间。
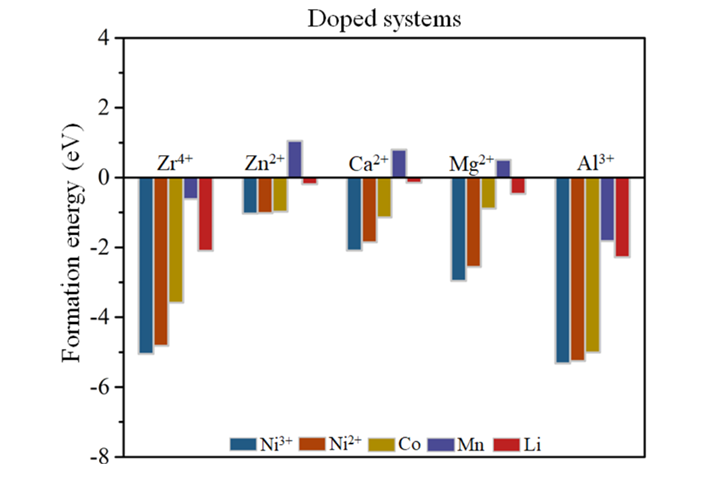
通过计算掺杂形成能,揭示了各掺杂元素在NCM811中的优先占位倾向。结果表明,所有掺杂元素(Zr⁴⁺、Zn²⁺、Ca²⁺、Mg²⁺、Al³⁺)均倾向于占据不稳定的Ni³⁺位点,而非Ni²⁺、Co、Mn或Li位点。这一发现具有重要意义,因为Ni³⁺的迁移是导致Li/Ni混排和结构退化的主要原因。
通过占据Ni³⁺位点,这些掺杂元素不仅减少了Ni³⁺的数量,还通过其自身的化学特性抑制了Ni³⁺向Li层的迁移。例如,Zr⁴⁺和Ca²⁺的高价态和较大离子半径能够有效“锚定”晶格结构,减少Li层塌陷的风险。这种占位偏好为后续研究掺杂对材料性能的影响提供了理论依据。
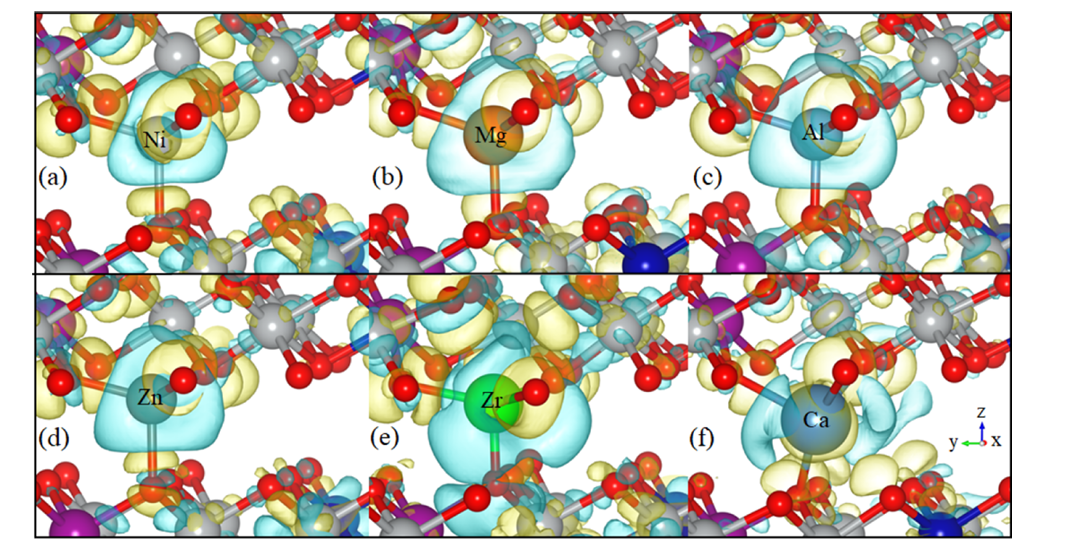
通过电荷密度差分析,进一步揭示了掺杂元素与氧原子之间的电子相互作用。结果显示,Ni³⁺、Mg²⁺、Al³⁺和Zn²⁺与O之间主要表现为离子键,其中Al³⁺和Mg²⁺的电荷转移更为显著,表明它们与O的键合更强。这种强键合虽然增强了局部结构的稳定性,但也可能导致Li层间距收缩,从而阻碍Li⁺的扩散。
相比之下,Zr⁴⁺和Ca²⁺的电荷密度分布显示出部分电子排斥效应,这种特性能够有效抵消Li层在脱锂过程中的收缩趋势,维持层间距离,为Li⁺的快速迁移创造条件。这种电子结构的差异直接影响了材料的电化学性能,尤其是循环稳定性和倍率性能。
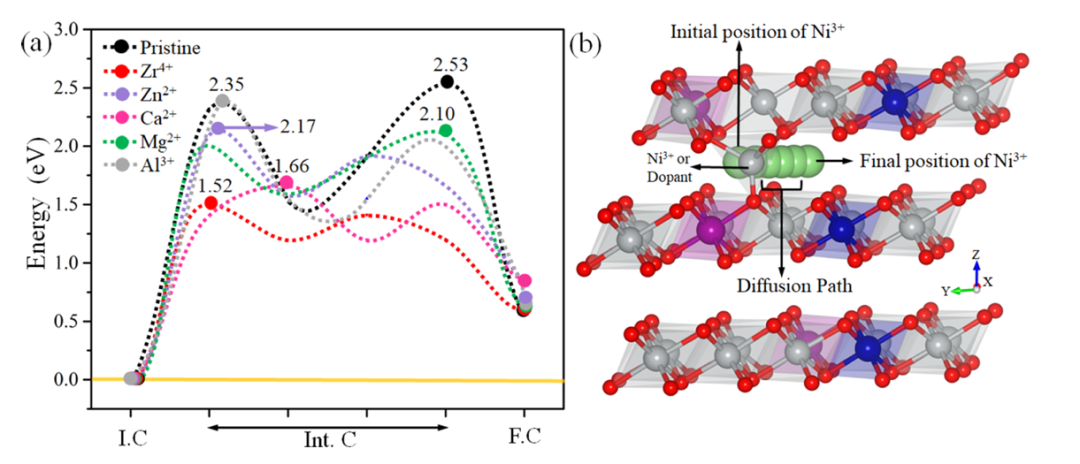
通过计算Li⁺在掺杂NCM811中的扩散能垒,定量评估了不同掺杂对Li⁺迁移动力学的影响。结果显示,Zr⁴⁺和Ca²⁺掺杂体系的Li⁺扩散能垒较低(分别为1.52 eV和1.66 eV),显著低于未掺杂的NCM811(2.53 eV)。
这一结果与键长和电荷密度分析一致,表明Zr⁴⁺和Ca²⁺的“柱撑效应”能够有效维持Li层间距,降低Li⁺迁移的阻力。相比之下,Al³⁺掺杂体系的Li⁺扩散能垒较高(2.35 eV),这与Al³⁺–O键的强键合特性及其导致的Li层收缩密切相关。
这些发现为优化富镍正极材料的倍率性能提供了重要指导,表明通过选择合适的掺杂元素(如Zr⁴⁺或Ca²⁺),可以显著提升Li⁺的迁移效率,从而改善电池的高倍率性能。
下图结合形成能和体积收缩分析,全面评估了掺杂NCM811在Li脱嵌过程中的结构稳定性。结果表明,Zr⁴⁺和Ca²⁺掺杂体系在脱锂过程中表现出较低的形成能和较小的体积收缩率(约4%),说明其结构稳定性显著优于未掺杂材料。
这种稳定性源于掺杂元素的“柱撑效应”,即在脱锂过程中,Zr⁴⁺或Ca²⁺迁移至Li层并占据空位,形成稳定的支撑结构,抑制了层间塌陷和颗粒破碎。
此外,电压曲线分析显示,Zr⁴⁺掺杂体系的电压平台更高,表明其具有更好的电化学可逆性和能量密度。这些计算结果与实验结果高度吻合,例如Zr⁴⁺掺杂NCM811在实验中表现出优异的循环稳定性和容量保持率。
综合来看,这些DFT计算不仅深入揭示了掺杂元素对NCM811结构稳定性和Li⁺扩散动力学的影响机制,还为实验设计高性能富镍正极材料提供了理论指导。
通过选择合适的掺杂元素(如Zr⁴⁺或Ca²⁺),可以有效抑制Li/Ni混排、维持层间结构稳定性、降低Li⁺扩散能垒,从而显著提升电池的循环寿命和倍率性能。
此外,研究还发现Al³⁺虽然能够增强局部键合强度,但会阻碍Li⁺扩散,因此在设计掺杂策略时需要权衡其利弊。
这些理论预测与实验观察的一致性,进一步验证了DFT计算在材料设计中的重要作用,为开发下一代高能量密度、长寿命锂离子电池正极材料提供了科学依据。
#华算科技 #富锂锰基氧化物 #NMC结构 #锂离子电池 #正极材料 #DFT计算 #锂离子扩散 #材料设计 #新能源电池技术 #结构稳定性 #能源存储 #LiMO2
? 点击阅读原文,立即下单!?