本文系统介绍了晶体掺杂的基本概念及其在半导体和催化领域的重要意义。掺杂通过引入外来杂质原子改变基质材料的物理化学性质,从而实现对材料性能的精准调控。
在半导体中,掺杂可调节载流子浓度和导电类型,为电子器件设计奠定基础;在催化领域,掺杂能优化活性位点的电子结构和表面性质,提升催化性能。
文章详细分析了替代式和间隙式两种常见掺杂模型的特点及其对材料结构和电子性质的影响。通过掺杂,可以显著改变材料的电学、光学和催化性能,为功能材料的设计与开发提供了重要手段。
晶体掺杂的基本概念
在材料科学中,晶体结构的掺杂是指通过引入外来杂质原子来改变基质材料物理化学性质的一种重要手段。从原子尺度来看,掺杂过程涉及杂质原子进入基质晶格并占据特定位置,这种原子替代或间隙占据会打破原有晶格的完美周期性,从而在电子结构和晶体场两方面产生显著影响。
根据杂质原子与基质原子的相对尺寸和电负性差异,掺杂可能导致晶格发生局部畸变或整体应变,这种结构变化往往与材料性能的调控直接相关。
在半导体中的意义
在半导体领域,向晶体结构中掺杂杂质元素是一项基础而关键的技术手段,其本质是通过可控引入外来原子来精确调控半导体的电学、光学和结构特性,从而实现对材料功能的定向设计。
从物理机制来看,掺杂过程打破了完美晶格的周期性势场,在禁带中引入杂质能级,这些能级可以成为载流子的来源或复合中心,从根本上决定了半导体的导电类型和载流子浓度。
对于元素半导体如硅、锗等,III族或V族元素的替代式掺杂可分别形成受主或施主能级,实现p型或n型半导体的制备,这是构建pn结、双极型晶体管等基础器件的前提。在化合物半导体领域,掺杂的复杂性更高,不仅需要考虑杂质能级的位置,还要关注掺杂元素在不同亚晶格中的占位倾向及其对化学计量比的影响。
从能带工程的角度,适当的掺杂可以调节半导体的费米能级位置,改变载流子的有效质量,甚至诱导能带结构的拓扑变化,这些效应在新型电子和光电器件设计中至关重要。
在光电特性调控方面,特定杂质元素的引入可以显著改变半导体的光吸收边、发光效率和载流子寿命,这对于太阳能电池、发光二极管和光电探测器等应用具有决定性影响。
某些深能级掺杂虽然会降低多数载流子浓度,但能通过陷阱辅助机制调控载流子的复合动力学,在特定器件中发挥积极作用。从材料稳定性角度看,精心设计的掺杂可以抑制晶格缺陷的迁移和增殖,提高半导体材料在高温、高场或辐照等恶劣环境下的工作可靠性。
现代半导体技术已发展出多种精确控制掺杂浓度分布的工艺方法,如离子注入、扩散和外延掺杂等,这些技术能够实现从轻掺杂衬底到重掺杂接触区的梯度过渡,满足复杂器件结构的需求。
随着半导体器件向纳米尺度发展,掺杂的微观不均匀性和统计涨落效应变得不容忽视,这促使研究者深入探索掺杂原子在极限尺寸下的分布规律和电学行为。在宽禁带半导体和低维半导体材料中,掺杂机制往往表现出与传统体材料不同的特征,需要建立新的理论模型进行描述。

在催化领域的意义
在催化领域,向晶体结构中掺杂杂质元素是一种关键的催化剂设计策略,其核心在于通过原子尺度的结构调控来优化催化性能。这种掺杂过程通过引入外来原子改变基质材料的本征特性,从而实现对催化剂电子结构、表面性质和活性位点的精准调控。
从本质上讲,掺杂可以视为一种“原子工程“手段,通过打破原有晶格的对称性和周期性,在材料中创造新的电子态和活性中心。在电子结构层面,掺杂能够调节催化剂的能带结构和费米能级位置,改变关键活性位点的电子密度分布,进而影响反应中间体的吸附强度和活化能垒。
特别是对于过渡金属基催化剂,掺杂可显著改变其d带中心位置,这一参数直接决定了催化剂表面对反应物分子的吸附能力。同时,杂质原子的引入往往会在晶格中产生应变效应和缺陷结构,这些结构畸变虽然可能破坏长程有序性,但却能在原子尺度创造具有特殊配位环境的活性位点,这些位点通常表现出不同于完美晶面的催化活性。
在催化稳定性方面,适当的掺杂能够增强材料的结构鲁棒性,抑制活性组分在使用过程中的烧结和流失,这对高温催化过程尤为重要。某些特定的掺杂元素还能阻断毒化物种与活性中心的结合,提高催化剂的抗中毒能力。
从作用机制来看,掺杂效应不仅取决于杂质元素本身的特性,更与它在基质晶格中的化学状态和局部环境密切相关。同一种掺杂元素在不同基质中可能呈现不同的价态和配位形式,这使得掺杂催化体系展现出丰富的构效关系。
现代催化研究特别关注掺杂引起的协同效应,即通过多种元素的共掺杂产生超越单一组分简单加和的催化性能。随着表征技术的发展,研究者已能在原子尺度解析掺杂位点的精细结构,并建立其与催化活性的直接关联。
理论计算和机器学习方法的引入进一步加速了新型掺杂催化剂的理性设计,使得人们能够预测最优的掺杂组合和配置方式。
总体而言,晶体掺杂作为一种灵活的材料改性策略,为开发高效、稳定、选择性的催化剂提供了广阔空间,其科学内涵和应用价值仍在不断拓展和深化中。
常见的掺杂晶体模型
替代式掺杂模型
替代式掺杂模型中,杂质原子占据了晶体中原本基体原子的晶格位置。下图中展示了不同元素(B、C、N、Al、P)掺杂对硅烯几何结构和电子性质的调控作用,其意义在于揭示了单原子掺杂如何通过改变材料的物理化学性质来优化其电催化性能。
从几何结构来看,掺杂原子的引入显著影响了硅烯的键长和形貌。B、C、N等较小原子导致Si-X键长缩短(Si-B为1.94 Å,Si-C为1.83 Å),而Al和P的键长接近原始Si-Si键长(2.23 Å)。
这种键长变化不仅引发局部晶格畸变,还导致硅烯平面发生弯曲(C@Si弯曲至170°,N@Si弯曲至155°),从而改变了材料的表面形貌和活性位点分布。
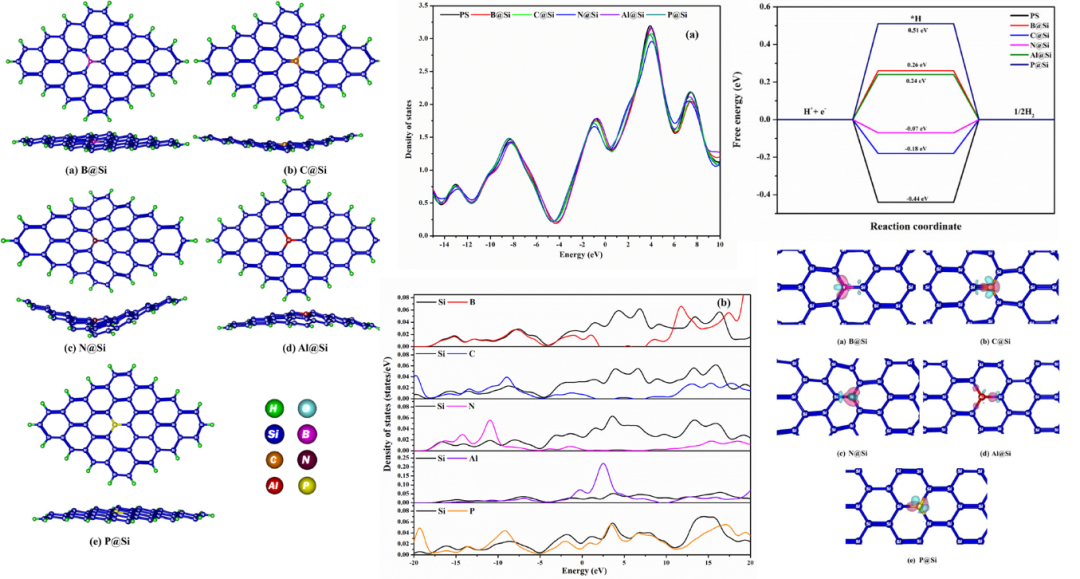
在电子性质方面,态密度(DOS)和投影态密度(PDOS)分析显示,不同掺杂原子对硅烯的能带结构产生了显著影响。B掺杂在未占据区引入了新能级,可能增强材料的电子接受能力;C和N掺杂则在占据区形成新能级,特别是N掺杂由于强电负性使能级靠近费米能级,显著减小了带隙(N@Si带隙降低21%),从而提升了电导率和反应活性。
Al掺杂在未占据区引入少量能级,而P掺杂在占据和未占据区均有贡献,导致带隙变化(P@Si带隙降低21%)。
此外,电子密度差图和bader电荷分析表明,B、C、N掺杂原子带负电(如N为-1.645 e),说明电荷从Si转移到掺杂原子,增强了局域亲核性;而Al和P掺杂原子带正电(如Al为+0.632 e),可能促进亲电反应。
这些电子和结构变化直接影响了硅烯的催化性能。在氢析出反应(HER)中,N@Si表现出最佳的催化活性,其ΔGH*(-0.07 eV)接近理想值,过电位低至70 mV,优于Pt(90 mV),这归因于N掺杂优化了氢吸附自由能和表面电荷分布。C@Si(过电位180 mV)和B@Si(ΔGH* 0.26 eV)也表现出良好的HER活性。
在氧析出反应(OER)中,C@Si的过电位最低(1.55 V),因为C掺杂平衡了中间体(OH、O)的吸附强度;P@Si(1.64 V)则因电荷转移特性对OOH*吸附有利。然而,掺杂对氧还原反应(ORR)的提升有限,这可能与掺杂后材料亲核性增强不利于氧还原步骤有关。
间隙式掺杂模型
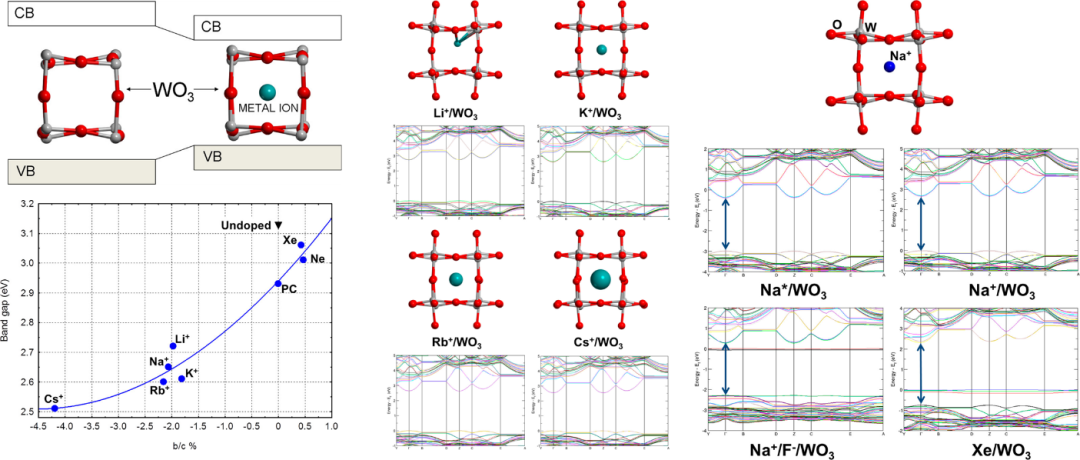
在间隙式掺杂模型中,杂质原子并不替代晶体中的基体原子,而是位于晶格原子之间的间隙位置。下图展示了Li⁺、K⁺、Rb⁺和Cs⁺等碱金属离子间隙掺杂对WO₃结构和电子性质的影响,其意义不仅在于揭示了掺杂对材料性能的调控机制,还为设计新型功能材料提供了重要依据。
从结构角度来看,碱金属离子的引入导致WO₃晶格发生各向异性畸变,具体表现为沿(001)方向的c参数显著增加,而b参数收缩,这种变化类似于从单斜相向四方相的局部结构转变。
这种畸变程度与掺杂离子的尺寸密切相关,例如Li⁺引起的体积变化较小(-0.4%),而Cs⁺则导致3.2%的体积膨胀,说明离子半径越大,对晶格结构的扰动越显著。
值得注意的是,这种结构变化并非简单的体积膨胀,而是通过改变晶胞参数的比例(如b/c比值)来影响材料的整体性质。在电子性质方面,所有碱金属离子掺杂均显著降低了WO₃的带隙,其中Li⁺使带隙减小0.4 eV,而Cs⁺的减小幅度达到0.6 eV。
这种带隙调控并非源于单纯的电荷注入或静电效应,因为对比实验表明,中性原子(如Xe)虽然引起类似的体积膨胀,却几乎不影响带隙值。此外,通过点电荷模型和H⁺掺杂的验证发现,纯电静作用仅贡献约0.2 eV的带隙减小,进一步证实结构畸变在带隙调控中的主导作用。
这种结构-性能关系的具体表现为:当b/c比值偏离纯WO₃的基准值时,带隙随之减小,且偏离程度越大,带隙减小越显著。这一发现为理解N₂分子掺杂WO₃导致带隙降低的争议提供了新视角,说明几何畸变可能是此类现象的共同机制。
从应用层面看,这项研究为优化WO₃的光催化性能指明了新方向。通过选择合适的碱金属离子进行间隙掺杂,可以精确调控材料的带隙宽度,使其光吸收边红移至可见光范围,从而显著提升太阳能转化效率,特别是在光催化水分解和光电化学电池等领域的应用潜力。
此外,研究还揭示了低浓度掺杂与高浓度“钨青铜“形成之间的过渡行为,为开发具有梯度功能的智能材料提供了理论支撑。