



DOI: 10.1021/acs.chemrev.0c00608
掺杂是指在材料(如半导体或氧化物)中引入少量杂质原子,以调控其电子结构、导电性和光学性质的过程。这些杂质可作为施主或受主,改变载流子浓度和能带结构;在量子材料中,掺杂往往诱导相变或超导行为;在能源材料中,掺杂可提升催化活性和离子传输。
掺杂的核心意义源于缺陷工程理论,其中杂质原子稳定局部电子态,降低能带隙或产生新能级,从而实现材料性能优化。传统实验方法如Hall效应测量可表征掺杂效应,但理论计算方法在揭示原子级机制和预测性能方面具有独特优势。
这些计算工具不仅能评估掺杂形成能,还能模拟其对材料热、电和光学性质的影响,推动从掺杂设计到应用优化的材料创新。
理论计算在掺杂研究中扮演关键角色,用于预测掺杂形成能、电子结构变化和性能优化。以下介绍主要计算方法及其在掺杂中的应用。
1、密度泛函理论(DFT)
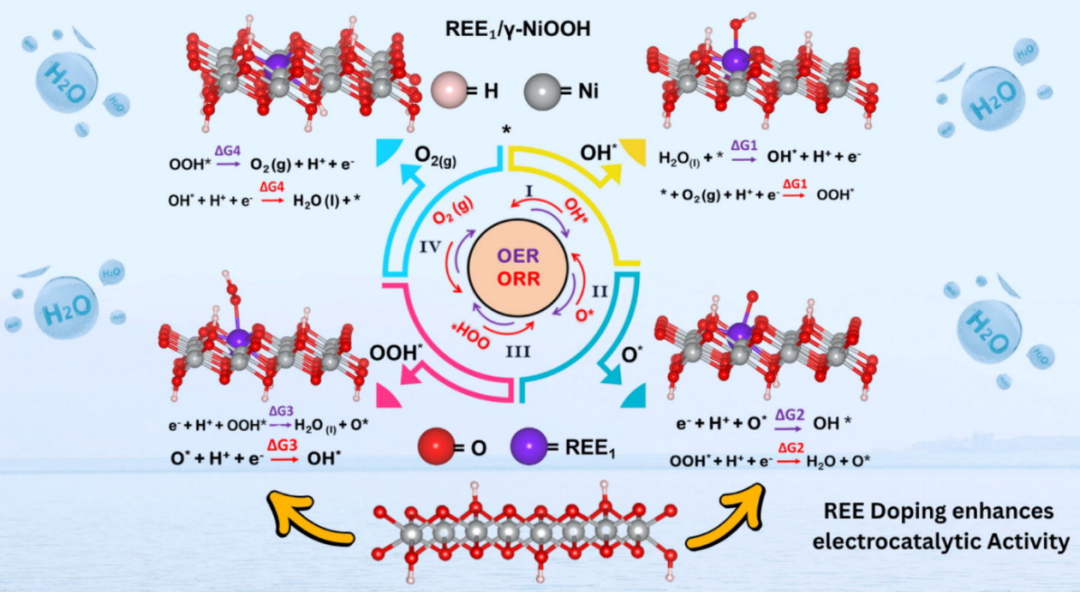
DOI: 10.1016/j.jechem.2025.07.003
密度泛函理论基于量子力学,计算掺杂材料的电子结构、形成能和反应势垒,是研究掺杂最常用的方法。其核心优势是无需经验参数,直接从电子密度层面预测杂质诱导的能带变化和电子转移。
例如,自旋极化密度泛函理论(DFT)研究了锚定稀土元素对γ-NiOOH 晶格表面的影响,旨在确定析氧反应(OER)和氧还原反应(ORR)的最佳催化位点。在通过 Ni 空位识别出合适的活性位点后,引入稀土元素(REE1)作为单原子催化(SAC)的掺杂剂。
2、分子动力学(MD)
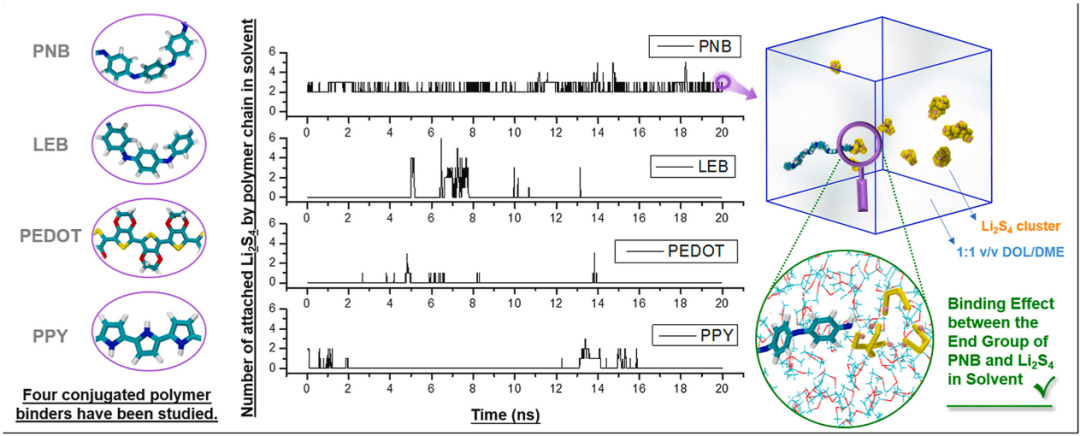
分子动力学通过经典力学模拟原子运动,研究掺杂材料的动态行为和有限温度效应,适用于大尺度体系的动态分析。其核心优势是能够捕捉掺杂原子在晶体中的扩散路径和热力学行为,尤其在高温或非平衡条件下。
例如,MD表明PNB的端基2可以有效地结合四硫化锂(即Li2S4)簇或43 Li2S4中的2个具有溶剂作用的分子。
但是,PNB,LEB,PEDOT和PPY的重复单元似乎无法通过非键相互作用来键合溶剂化的Li2S4,特别是当阴极局部区域中的四硫化物/粘合剂的浓度较低时。
因此,具有该特定官能团的聚合物(建议进一步研究PNB的端基2)作为抑制溶剂化多硫化锂穿梭效应的潜在有效粘合剂。
3、机器学习
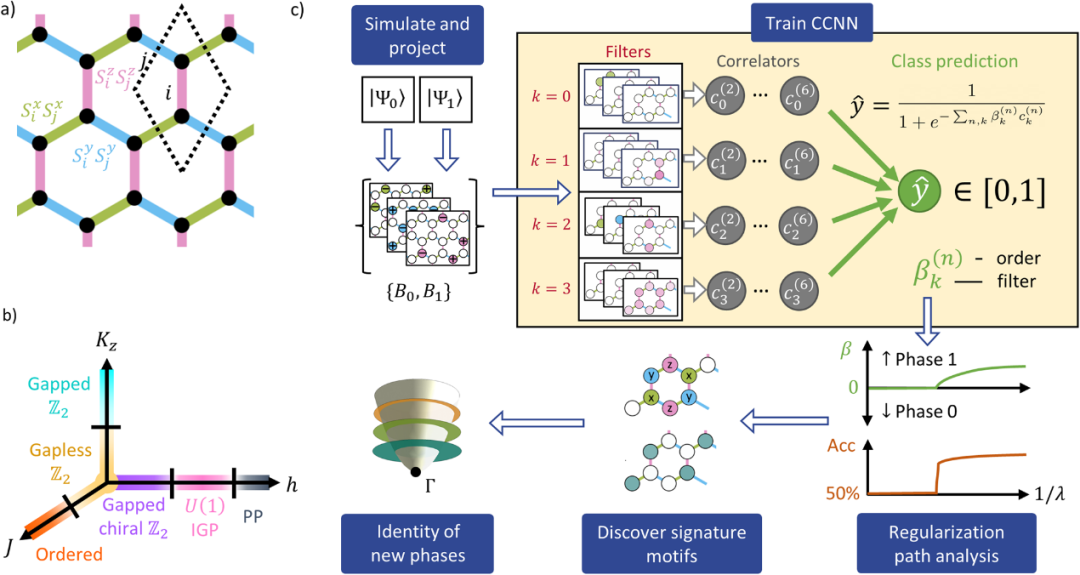
DOI: 10.1038/s42005-024-01542-8
机器学习通过数据驱动方法,优化掺杂材料的计算效率和预测精度,特别适合高通量筛选和复杂体系分析。其核心优势是从DFT或实验数据中学习特征,快速预测掺杂形成能和性质。
例如,利用可解释的经典机器学习挖掘采样投影快照的量子经典混合方法(QuCl)可以揭示看似无特征的量子态的特征。外部磁场下蜂窝晶格的 Kitaev-Heisenberg 模型提供了一个测试 QuCl 的理想系统,其中模拟发现了夹在已知相之间的中间无间隙相 (IGP),引发了对其难以捉摸的性质的争论。
我们使用相关器卷积神经网络,在标记的投影快照上进行训练,结合正则化路径分析来识别阶段的特征,证明 QuCl 再现了已建立相的已知特征。
掺杂作为材料调控的核心,通过引入杂质实现电子结构和性能优化,成为材料科学和能源领域的焦点。密度泛函理论、分子动力学和机器学习通过电子结构建模、动态模拟和数据驱动预测,为掺杂的机制解析和优化提供了强大支持。
这些方法显著推进了半导体、催化剂和能源材料中的研究。随着计算技术和算法的进步,如机器学习与DFT的深度整合,掺杂的设计将进一步加速,为可持续能源和先进材料提供新机遇。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???