

氧还原反应(ORR)是燃料电池和金属–空气电池中关键的电化学反应之一,其催化性能直接影响电池的效率和寿命。近年来,密度泛函理论(DFT)计算在ORR机制研究中发挥了重要作用,为理解催化剂的活性位点、反应路径和能量势垒提供了理论支持。以下将结合多篇文献中的DFT计算结果,详细分析ORR的常见DFT计算结果。



在ORR过程中,氧气分子(O₂)在催化剂表面被还原为水(H₂O)或过氧化氢(H₂O₂),具体取决于反应机制和催化剂类型。DFT计算通常用于确定ORR的反应路径,并计算各中间体的吸附能和自由能变化。
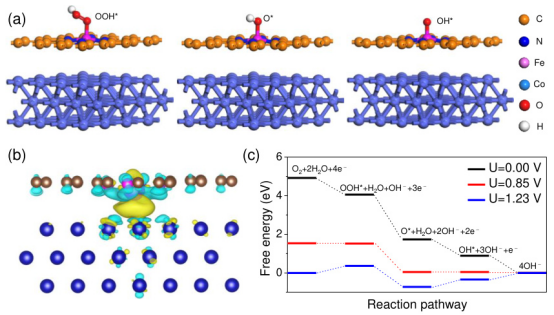
例如,Co@Fe-N-C催化剂在ORR中的DFT计算结果图(a)显示了反应过程中三种中间体(OOH*、O和OH)的简化结构图,这些中间体在催化剂表面参与ORR反应。
图(b)则显示了Co@Fe-N-C催化剂的电荷密度差分布,通过颜色变化直观地反映了电荷分布的变化。图(c)为DFT计算得出的自由能图,展示了从氧气(O₂)到最终产物(OH⁻)的反应路径上的自由能变化。不同电压(U=0.00 V、U=0.85 V、U=1.23 V)条件下,各中间体的自由能变化情况。
这些结果表明,Co@Fe-N-C催化剂在不同电压下表现出优异的ORR活性。
催化剂的表面结构和电子性质对ORR的活性有显著影响。通过DFT计算研究了CoO表面不同晶面的原子结构与ORR/OER活性之间的关系表明,{111}-Ov晶面具有最佳的ORR/OER活性,其电子结构优化了中间体的吸附和电荷转移,从而提高了整体电催化活性。实验结果证实了理论预测,表明原子结构工程可以成功增强ORR/OER活性。
配位环境对ORR中间体的吸附能和反应路径有重要影响。原子分散的Mn-(N-C₂)₂(O-C₂)₂位点催化ORR的机理表面通过实验确定的Mn-SAs配位结构,构建了Mn-(N-C₂)₂(O-C₂)₂和Mn-(N-C₂)₄的结构模型。
计算结果显示,Mn-(N-C₂)₂(O-C₂)₂位点上的中间体吸附自由能比Mn-(N-C₂)₄位点上的吸附自由能大得多,表明N和O的杂化配位降低了吸附能垒。对于Mn-(N-C₂)₂(O-C₂)₂催化ORR,所有中间步骤都是放热的,而对于Mn-(N-C₂)₄催化ORR,最后一个中间步骤是吸热的。
这些结果表明,改变Mn-SAs的配位环境可以显著增强ORR活性。
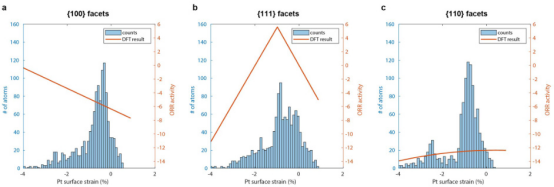
表面应变对催化剂的电子结构和反应活性有重要影响。不同晶面({100}, {111}, {110})的铂(Pt)表面应变与ORR活性之间的关系。图a、b、c分别对应{100}、{111}和{110}晶面。
每个子图中,左侧纵轴表示Pt表面应变的百分比,右侧纵轴表示ORR活性。蓝色柱状图代表实际测量的Pt表面应变分布,橙色曲线代表基于DFT计算的预测结果。
从图中可以看出,对于{100}晶面,随着Pt表面应变的增加,ORR活性逐渐降低;对于{111}晶面,ORR活性随着Pt表面应变的增加先增加后减少;而对于{110}晶面,ORR活性随着Pt表面应变的增加逐渐降低。这些结果表明,表面应变对ORR活性有显著影响。
离子在电极表面的积累和排斥对ORR活性有重要影响。研究人员研究了钾离子(K⁺)对酸性溶液中2电子ORR的电催化增强效应。通过原位X射线光电子能谱(XPS)测量,发现K⁺在碳电极表面的积累和排斥与施加的电位有关。
DFT计算表明,K⁺诱导的局部电场可以稳定2e⁻ ORR的关键中间体*OOH,从而提高反应活性。实验结果和理论计算结果一致,为2e⁻ ORR的反应机制提供了新的见解。
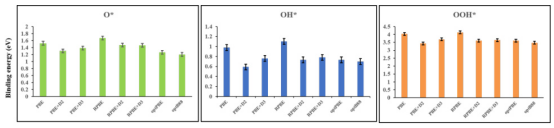
DFT方法的选择对ORR计算结果有重要影响。不同DFT方法(如PBE、PBE-D3、PBE-D1、RPBE、RPBE-D3、RPBE-D1、opPBE和opPBE-D3)对O*、OH和OOH中间体的吸附能的计算结果包括八种DFT方法对这些中间体的吸附能的计算结果。
每个柱状图代表一种DFT方法,柱状图的高度表示吸附能的大小,单位为电子伏特(eV)。图中显示,对于O*、OH和OOH中间体,不同DFT方法的计算结果存在差异,但总体趋势较为一致。例如,对于O*中间体,PBE-D3方法的吸附能最高,而opPBE-D3方法的吸附能最低。这些结果表明,DFT方法的选择对ORR计算结果有重要影响。
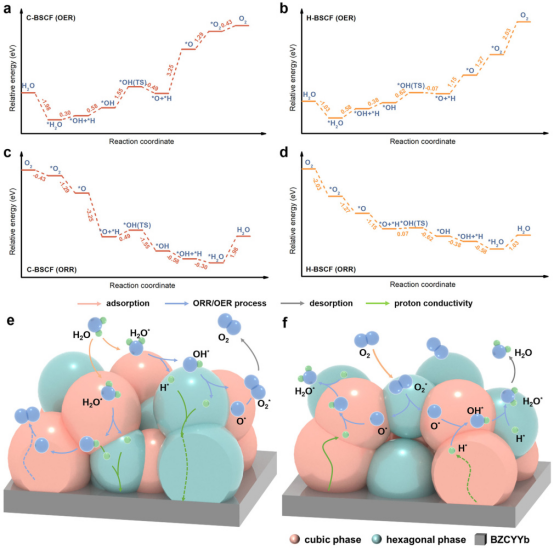
新型催化剂的DFT计算为ORR研究提供了新的思路。密度泛函理论(DFT)计算结果显示探讨了在R-PECs空气电极上过氧化氢还原反应(ORR)和氧气析出反应(OER)的机制。
图a和b分别显示了在立方C-BSCF(100)和六方H-BSCF(0001)表面进行OER过程的能量势垒。图c和d则展示了在立方C-BSCF(100)和六方H-BSCF(0001)表面进行ORR过程的能量势垒。图e和f则详细描绘了在混合C/H-BSCF表面上的双相协同OER和ORR催化机制。
这些结果表明,新型催化剂在ORR和OER中表现出优异的催化性能。
过渡金属酞菁(TM-Pc)作为非贵金属催化剂在ORR中表现出良好的活性。通过DFT计算研究了单核和平面双核钴(CoPc)和铁酞菁(FePc)催化剂在ORR中的催化性能,计算结果表明,Fe基酞菁的ORR活性远高于Co基酞菁,这是因为前者可以催化4电子的ORR,而后者只能催化2电子的ORR。
Fe基酞菁的高活性可归因于其最高占据分子轨道(HOMO)的能量水平较高,这导致催化剂与ORR物种之间的吸附能更强。此外,与单核FePc相比,平面双核FePc在酸性介质中具有更稳定的结构和更适合ORR物种的吸附能,使其成为ORR的有希望的非贵金属电催化剂。
钙钛矿氧化物(LaBO₃)作为ORR催化剂在碱性电解质中表现出良好的活性。通过DFT计算比较了LaMnO₃、LaFeO₃和LaCrO₃三种材料的ORR活性发现,混合泛函方法计算的结果与实验观察到的ORR活性顺序(LaMnO₃ > LaCrO₃ > LaFeO₃)更一致,而GGA或GGA+U方法的结果则与实验结果不符。这些结果表明,不同DFT方法对ORR活性的预测结果存在差异,需要进一步优化计算方法。