

本文系统介绍了活性位点的定义及其在化学反应中的核心作用,重点探讨了通过多种理论计算方法识别和表征活性位点的策略。文章详细分析了态密度(DOS)和d带中心在揭示电子结构中的作用,吸附能与活性位点反应活性的关联,静电势对亲电/亲核位点的预测能力,以及HOMO-LUMO能级差对分子反应活性的影响。
此外,还介绍了福井函数和配位不饱和度在活性位点研究中的应用,结合具体案例(如过渡金属催化剂、有机分子反应位点)展示了这些方法在催化机制解析和材料设计中的实际价值。这些研究为理解催化反应机理、优化催化剂性能提供了理论工具和科学依据。



活性位点的定义与概念


活性位点,又称活化位置,是指在化学反应中,物质表面或分子结构中能够直接参与并主导反应进行的特定原子、原子团或分子区域。这些位点具有独特的电子结构和几何特征,使其能够与反应物分子发生特异性的相互作用,从而显著降低反应的活化能,促进化学反应的发生,在众多化学反应过程中扮演着核心角色。
在多相催化领域,活性位点同样发挥着关键作用。多相催化反应通常发生在催化剂表面,催化剂表面的原子或原子团构成了活性位点。这些活性位点能够吸附反应物分子,改变反应物分子的电子云分布和化学键状态,进而促进反应的进行。
通过性质判断活性位点位置
1. 态密度(DOS)
态密度(DOS)的概念与计算
态密度(Density of States,DOS)是凝聚态物理中描述电子结构的重要概念,它表示单位能量范围内的电子状态数目。态密度反映了电子在不同能量状态下的分布情况,是研究材料电子结构和物理性质的关键参数。在材料的电子结构中,态密度起着核心作用。
对于金属材料,其费米能级(EF)位于连续的电子态密度分布区域内,表明在费米能级附近存在大量可参与导电的自由电子,这使得金属具有良好的导电性。而对于半导体材料,费米能级处于价带和导带之间的禁带中,在绝对零度时,价带被电子完全占据,导带为空,随着温度升高或受到外界激发,电子可从价带跃迁到导带,产生导电载流子,其导电性能介于导体和绝缘体之间。
对于绝缘体,其禁带宽度较大,电子难以从价带跃迁到导带,因此在通常条件下几乎不导电。态密度的分布特征与材料的物理性质密切相关,不同材料的态密度曲线形状各异,反映了其独特的电子结构和化学键特性。
d带中心的含义与作用
在过渡金属及其化合物中,d带中心是一个重要的概念,它描述了d轨道电子能量的平均位置。d带中心的位置对材料的电子结构和化学性质有着深远的影响。d带中心与催化剂活性密切相关,这主要是基于Sabatier原理。Sabatier原理指出,对于一个催化反应,最佳的催化剂应该与反应物和反应中间体具有适中的相互作用强度。
当d带中心靠近费米能级时,材料对反应物分子的吸附能力增强,因为d带中心的位置决定了反键态的能量,d带中心越靠近费米能级,反键态能量越低,反应物分子与催化剂表面原子形成的化学键越强,吸附作用也就越强。
如果吸附过强,反应中间体难以脱附,会导致催化剂表面被占据,阻碍后续反应的进行,从而降低催化活性;反之,若d带中心远离费米能级,吸附作用过弱,反应物分子难以被活化,同样不利于催化反应的发生。
案例分析
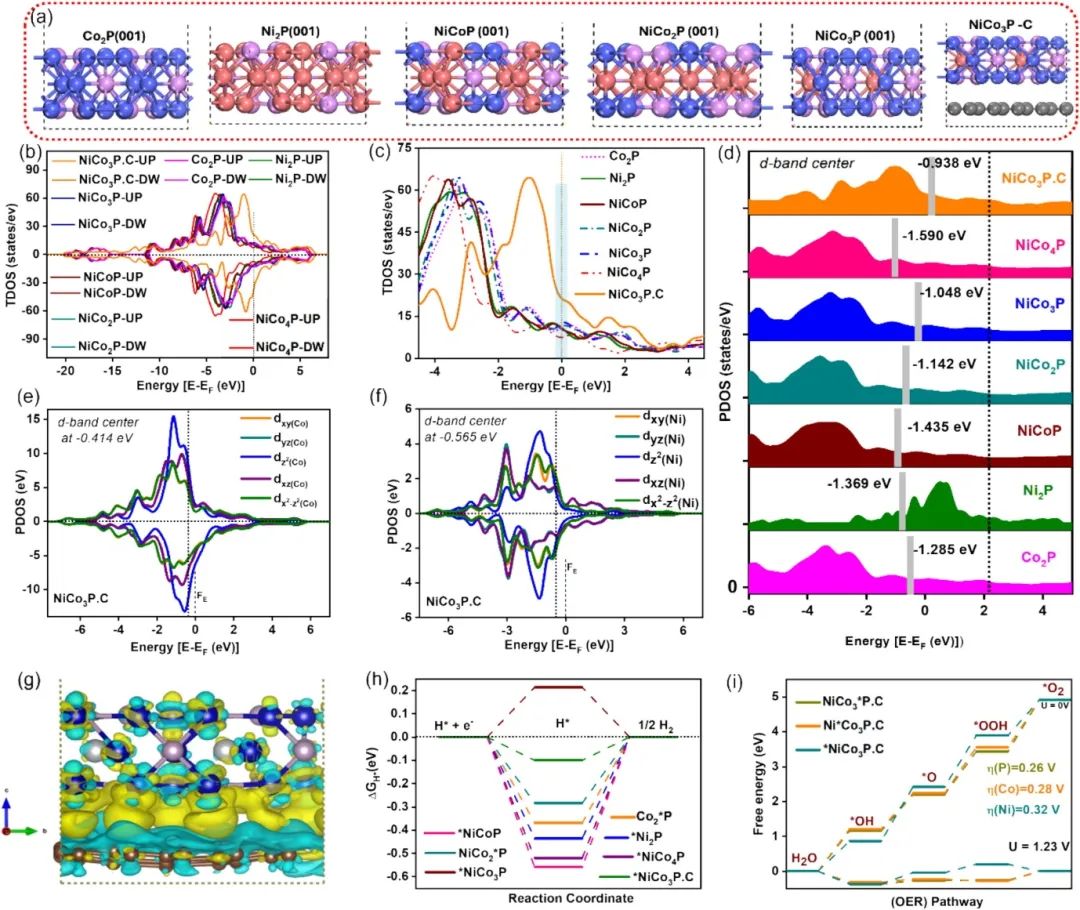
DOS可呈现材料电子态分布,以文中NiCo3P等磷化物为例,其DOS能反映费米能级(EF)附近电子态密度占比。若费米能级附近电子态密度高,说明该区域电子易参与反应,如NiCo3P异质结在自旋向上构型中,费米能级附近态密度显著增加,预示此处电子转移活跃,是反应潜在位点。
d带中心(εd)描述过渡金属d轨道电子分布能量位置,与催化剂吸附能力紧密相关。d带中心越靠近费米能级,催化剂对反应中间体(如HER中H*、OER中OOH*等)吸附作用越强。文中NiCo3P的d带中心(εd=-0.938eV)高于其他催化剂(如NiCo4P为-1.590eV),表明其对水吸附更易,利于后续催化反应。
d带中心上移,能优化催化剂d轨道与反应物轨道电荷转移,降低反应能垒:HER中使H*吉布斯自由能接近0eV,促进H2高效生成;OER中削减OOH*、O*、OOOH*等中间体能量壁垒,加速反应进程。
二者协同,DOS从电子态整体分布找活跃区域,d带中心精准刻画金属位点吸附特性,结合可明确电催化剂反应位点:通过分析DOS费米能级附近态密度,锁定电子转移关键区;依据d带中心判断位点对中间体吸附能力,筛选出高活性反应位点(如NiCo3P中Ni位点因d带中心及吸附能优势,成为HER关键位点),为设计高效电催化材料、优化反应路径提供核心支撑,助力理解催化机制与开发高性能催化剂。
2. 吸附能测试
吸附能的计算方法
吸附能是描述吸附过程中能量变化的重要物理量,它定量地衡量了吸附质与吸附剂之间相互作用的强弱。在基于密度泛函理论(DFT)的计算中,吸附能的计算公式通常表示为:Eads=Esystem-Esubstrate-Eadsorbate,其中Eads为吸附能,Esystem是吸附质吸附在吸附剂表面后整个体系的总能量,Esubstrate是吸附剂的能量,Eadsorbate是孤立吸附质的能量。
吸附能与活性位点的关系
吸附能的大小与活性位点对反应物的吸附能力紧密相关,进而对化学反应的活性产生重要影响。从本质上讲,吸附能反映了吸附质分子与活性位点之间相互作用的强度,这种相互作用包括静电相互作用、共价键作用、范德华力等多种形式。
当吸附能较大时,表明活性位点与反应物分子之间的相互作用较强,反应物分子能够更紧密地吸附在活性位点上。在催化反应中,这种较强的吸附作用有助于反应物分子的活化,使反应物分子的化学键发生变形或断裂,从而降低反应的活化能,促进反应的进行。
因此,活性位点对反应物的吸附能需要处于一个合适的范围,才能实现最佳的反应活性。通过调控活性位点的电子结构和几何构型,可以优化吸附能,提高反应活性。
案例分析
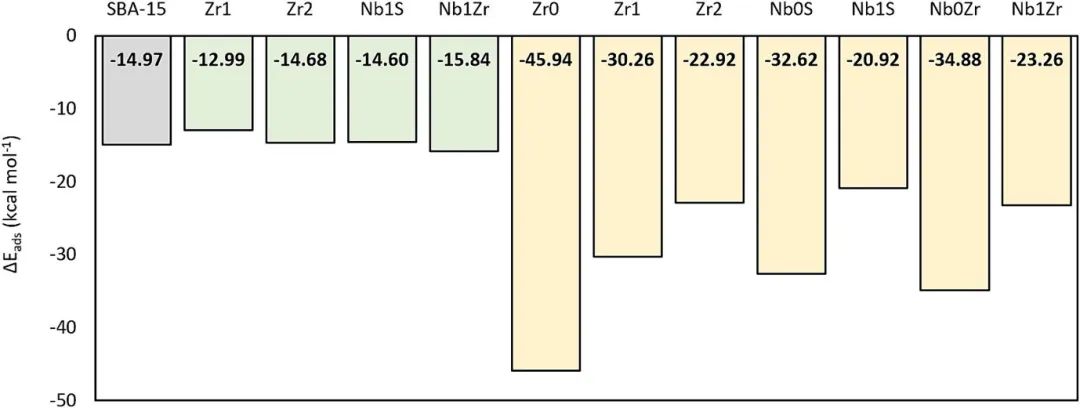
图中,氨吸附能(ΔEads)是确定反应位点酸性质的关键指标,借由不同颜色柱(灰、绿、黄分别对应硅醇基、M-OH簇、Lewis金属中心的参考值),可区分Brønsted酸位点与Lewis酸位点。对于ZrSBA-15和NbSBA-15体系,ΔEads能量化活性位点对氨的吸附作用强度,进而反映酸强度:值越负,吸附越强、酸强度越高。
如Zr0和Nb0S位点的ΔEads负值大,表明其Lewis酸位点活性高;随着金属中心连接OH基团数增加,ΔEads负值减小,反映位阻增大使酸强度降低,可据此确定不同OH连接情况下载体表面活性位点的反应活性顺序。
同时,对比ZrSBA-15与NbSBA-15的平均ΔEads,能判断两类材料整体酸强度差异(ZrSBA-15平均吸附能更负,酸强度更高),结合不同活性位点(如Zr、Nb不同取代位点)的ΔEads变化,可识别出对反应更具活性的特定位点,为探究催化反应中活性位点的作用、优化催化剂设计提供依据,助力明确哪些位点更利于吸附反应物、促进催化反应发生,精准锁定关键反应位点。
3. 静电势计算
静电势的原理与计算方法
静电势,从物理学角度而言,是指在静电场中,单位正电荷从无穷远处移动到某点时电场力所做的功,它是描述电场特性的一个重要物理量。在分子体系中,静电势反映了分子周围空间中电荷分布所产生的电场对其他电荷的作用。
静电势与活性位点的关联
静电势图能够直观地展示分子表面电荷的分布情况,这为确定活性位点的位置提供了重要线索。在静电势图中,颜色通常用于表示静电势的高低,红色区域表示静电势较低,即电子云密度较高,通常对应于富电子区域;蓝色区域表示静电势较高,即电子云密度较低,通常对应于缺电子区域。
而活性位点往往与静电势的极值区域相关。在亲电反应中,反应物中的亲电试剂倾向于进攻分子中静电势较低(富电子)的区域,因为这些区域具有较高的电子云密度,能够提供电子与亲电试剂发生反应。
在卤代烷烃的亲电取代反应中,卤素原子(如氯原子)具有较强的电负性,使得与卤素原子相连的碳原子周围电子云密度降低,静电势升高,成为缺电子区域;而卤代烷烃分子中的烷基部分相对富电子,静电势较低。
亲电试剂(如氢离子)更容易进攻烷基部分,从而发生亲电取代反应。在亲核反应中,情况则相反,亲核试剂倾向于进攻分子中静电势较高(缺电子)的区域。因此,通过分析静电势图中电荷分布的特点,可以预测分子在化学反应中可能的活性位点,为研究化学反应机理和反应活性提供重要依据。
案例分析:有机分子反应活性位点的确定
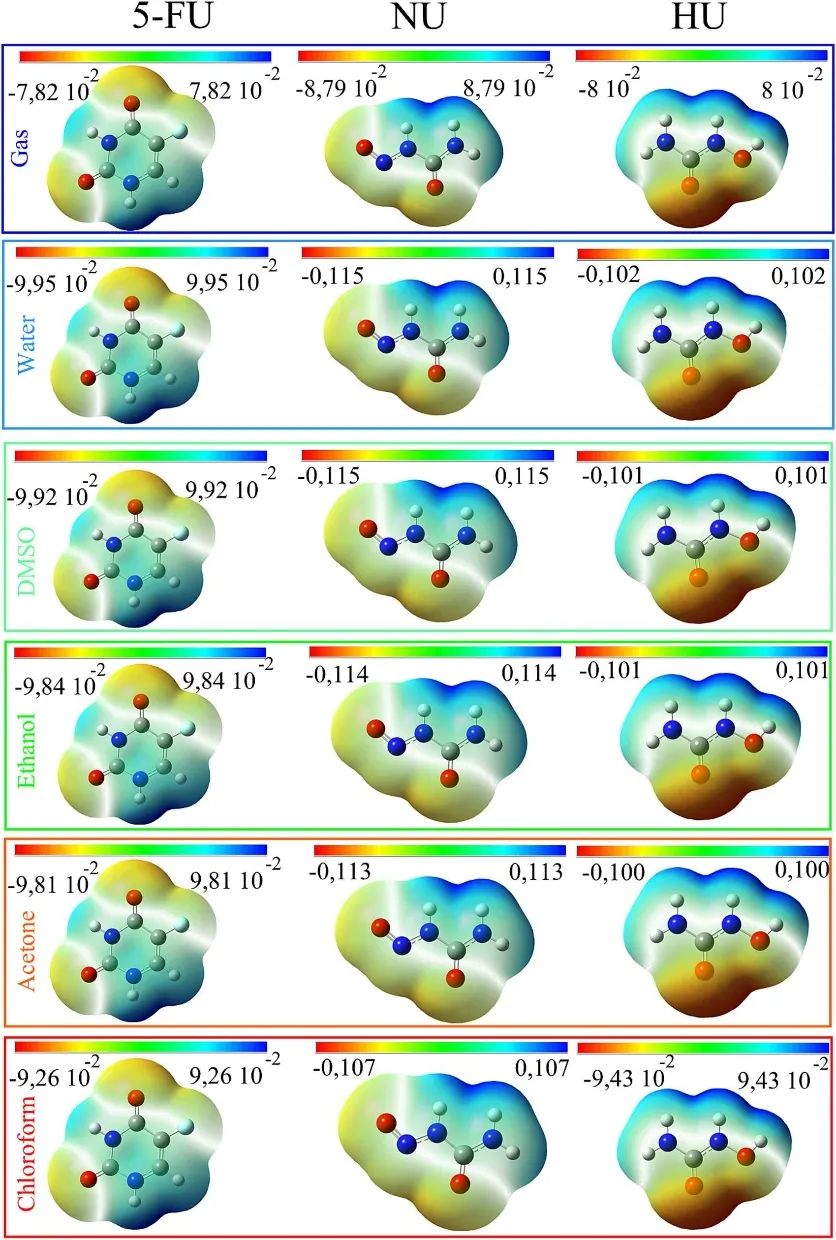
通过分析不同溶剂环境下的静电势分布,可以直观地识别分子中潜在的亲电和亲核反应位点。图中以颜色梯度(从红色到蓝色)表示静电势的变化:红色区域(如氧和氟原子周围)代表负静电势,表明这些位点容易受到亲电攻击(提供电子);蓝色区域(如氢原子周围)代表正静电势,表明这些位点容易受到亲核攻击(接受电子);而绿色和黄色区域则代表中性静电势,通常反应活性较低。这种可视化方法为理解药物与生物靶标或纳米粒子的相互作用机制提供了重要依据。
此外,溶剂极性对静电势分布的影响也被清晰地展现出来。随着溶剂极性的增加,静电势的绝对值显著增大(例如,5-FU的负静电势从7.82×10−2增至9.26×10−2,正静电势从8.79×10−2增至0.107),这表明在高极性环境中,抗癌药物的极性增强,反应位点的活性也随之提高。
值得注意的是,5-FU在气相和水相环境中的静电势分布变化较小,而NU和HU的变化更为显著,这与偶极矩的研究结果一致,进一步验证了溶剂效应对分子反应性的调控作用。
4. HOMO/LUMO
HOMO和LUMO的概念
最高占据分子轨道(Highest Occupied Molecular Orbital,HOMO)和最低未占据分子轨道(Lowest Unoccupied Molecular Orbital,LUMO)是分子轨道理论中的重要概念。
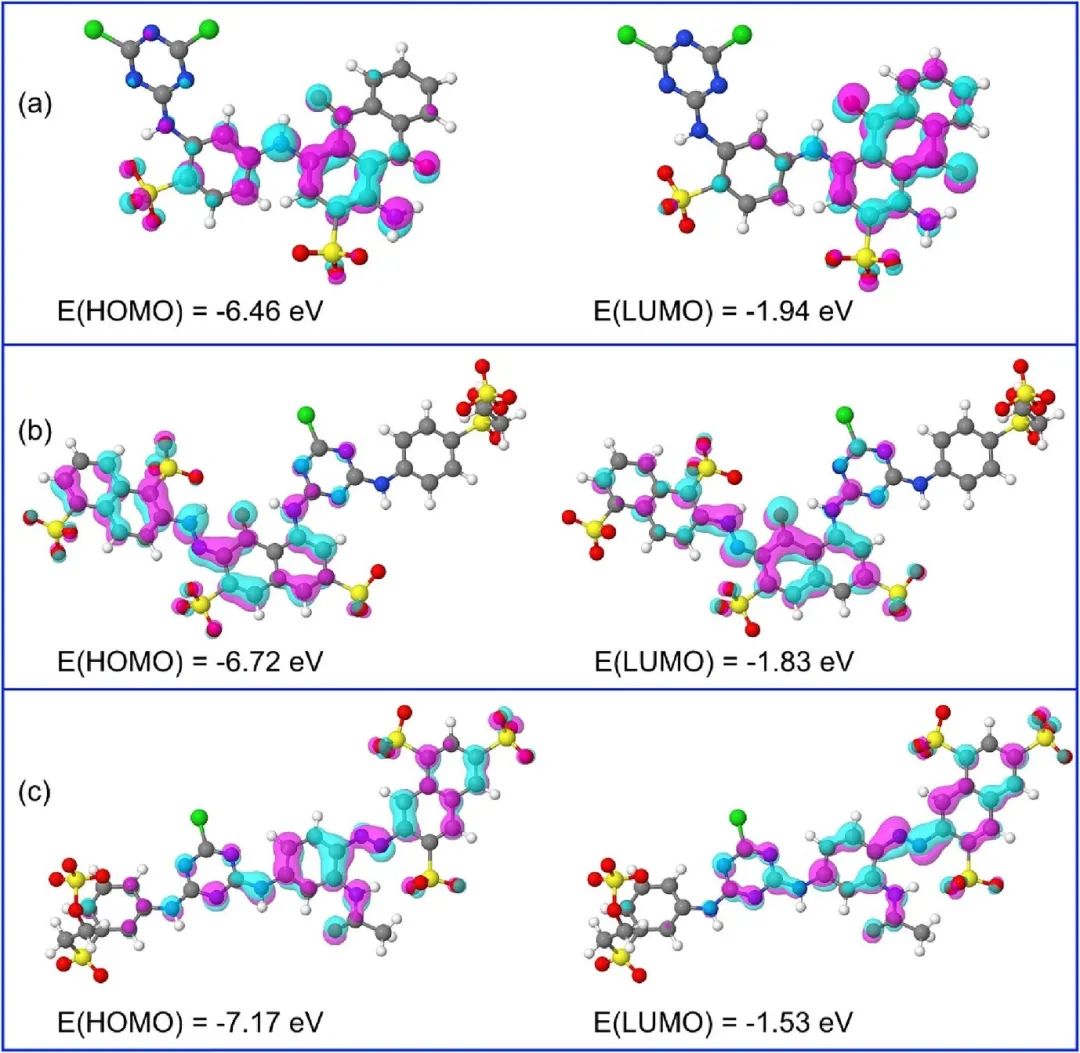
分子轨道理论认为,分子中的电子不再局限于某个原子的原子轨道上,而是在整个分子的范围内运动,分子轨道由原子轨道线性组合而成。HOMO是分子中能量最高的被电子占据的分子轨道,其中的电子具有相对较高的能量,相对较为活泼,容易参与化学反应,因为当分子与其他物质发生反应时,HOMO上的电子最有可能首先发生转移或参与成键。
LUMO则是分子中能量最低的未被电子占据的分子轨道,它对电子具有较强的吸引力,在化学反应中往往是接受电子的主要轨道。
HOMO/LUMO与反应活性的关系
HOMO/LUMO能级差(ΔE)对分子的反应活性起着关键作用。一般来说,能级差越小,分子越容易发生化学反应,因为较小的能级差意味着电子从HOMO跃迁到LUMO所需的能量较低,分子更容易被激发,从而参与反应。
在芳香族化合物中,苯的HOMO/LUMO能级差相对较大,使得苯具有较高的化学稳定性,不易发生加成反应,而更倾向于发生亲电取代反应。而对于一些具有共轭结构的分子,如1,3 -丁二烯,由于其共轭体系的存在,使得HOMO和LUMO的能量相对接近,能级差较小,因此1,3 – 丁二烯比苯更活泼,更容易发生加成反应。
HOMO和 LUMO的轨道分布也对反应选择性产生重要影响。不同的轨道形状和空间取向决定了分子在反应中与其他反应物分子相互作用的方式和位置。在亲电加成反应中,亲电试剂通常会进攻分子的LUMO,而亲核试剂则倾向于进攻分子的HOMO。
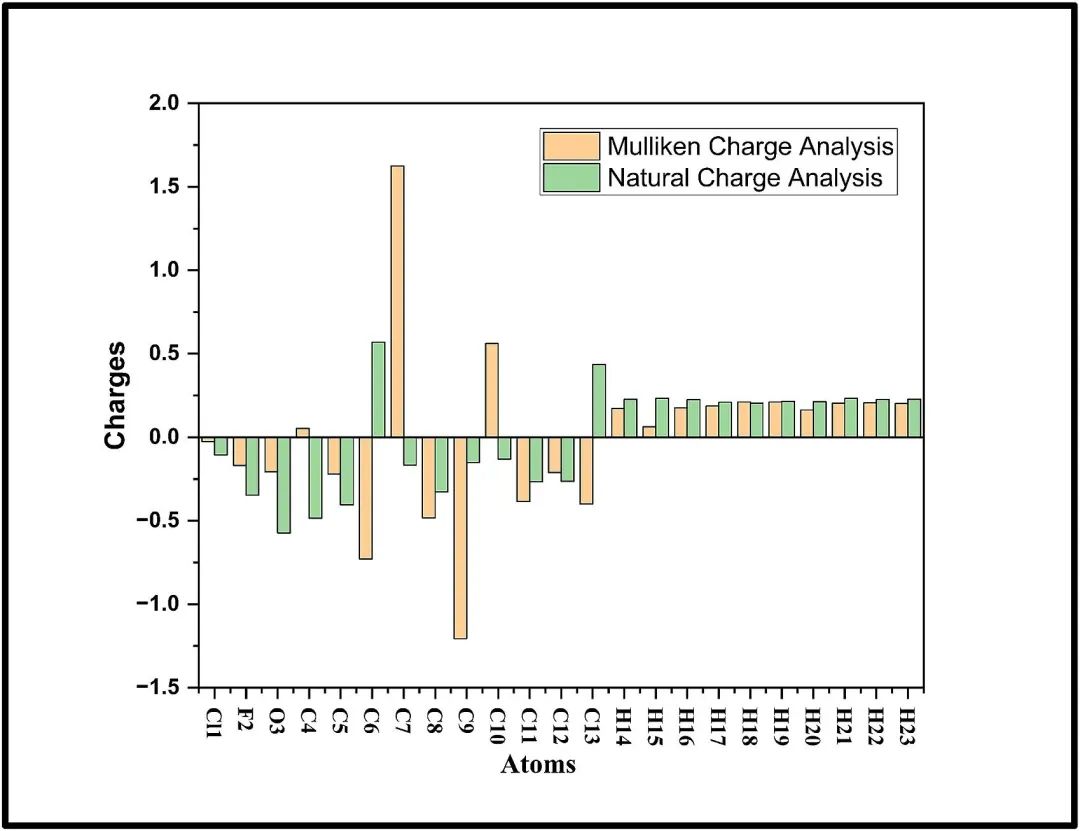
5. 电荷分布分析
电荷分布能够直观地反映原子的电子云密度,这与活性位点的反应活性密切相关。电子云密度的高低直接影响原子的化学活性,进而决定了活性位点在化学反应中的作用。
在化学反应中,电子云密度较高的原子具有较强的供电子能力,容易与亲电试剂发生反应;而电子云密度较低的原子则具有较强的吸电子能力,容易与亲核试剂发生反应。
在过渡金属催化剂中,活性位点的电荷分布对催化反应的活性和选择性起着关键作用。电荷分布在决定活性位点反应活性方面具有重要作用,通过合理调控电荷分布,可以优化材料在各种化学反应中的性能。
6. 福井函数
福井函数的定义与计算
福井函数(Fukui Function),又称前线电子密度,是由日本化学家福井谦一提出的一个重要概念,它在化学反应活性的研究中具有举足轻重的地位。福井函数基于分子轨道理论,用于描述分子中电子密度对原子的微小变化的响应,从而反映分子中不同原子的反应活性。
从理论基础来看,福井函数与分子的电子结构密切相关。在分子体系中,电子的分布决定了分子的化学性质和反应活性。福井函数通过量化电子密度的变化,为研究化学反应提供了一种微观层面的视角。福井函数的定义如下:对于一个分子体系,其电子数为N,当体系得到一个电子(电子数变为N+1)时,电子密度的变化量定义为亲核福井函数(f–);当体系失去一个电子(电子数变为N-1)时,电子密度的变化量定义为亲电福井函数(f+)。
福井函数与反应活性位点的预测
福井函数在预测分子的亲核、亲电反应位点以及反应活性方面具有重要作用。福井函数值的大小与原子的反应活性密切相关,福井函数值越大,表明该原子在化学反应中越容易参与反应,是潜在的活性位点。
在亲核反应中,亲核试剂通常会进攻分子中亲电福井函数(f+)值较大的原子,因为这些原子周围的电子云密度相对较低,呈现出缺电子特性,容易接受亲核试剂的电子对。
在卤代烷烃与氢氧化钠的亲核取代反应中,卤代烷烃分子中与卤素原子相连的碳原子的亲电福井函数值较大,是亲核试剂(氢氧根离子)进攻的活性位点。在亲电反应中,亲电试剂则倾向于进攻分子中亲核福井函数(f–)值较大的原子,这些原子周围电子云密度较高,具有较强的供电子能力,能够与亲电试剂发生反应。
通过计算福井函数,可以有效地预测分子在不同类型化学反应中的活性位点,为研究化学反应机理提供重要依据。福井函数还可以用于比较不同分子或同一分子不同异构体的反应活性。对于结构相似的分子,福井函数值较大的分子通常具有更高的反应活性。
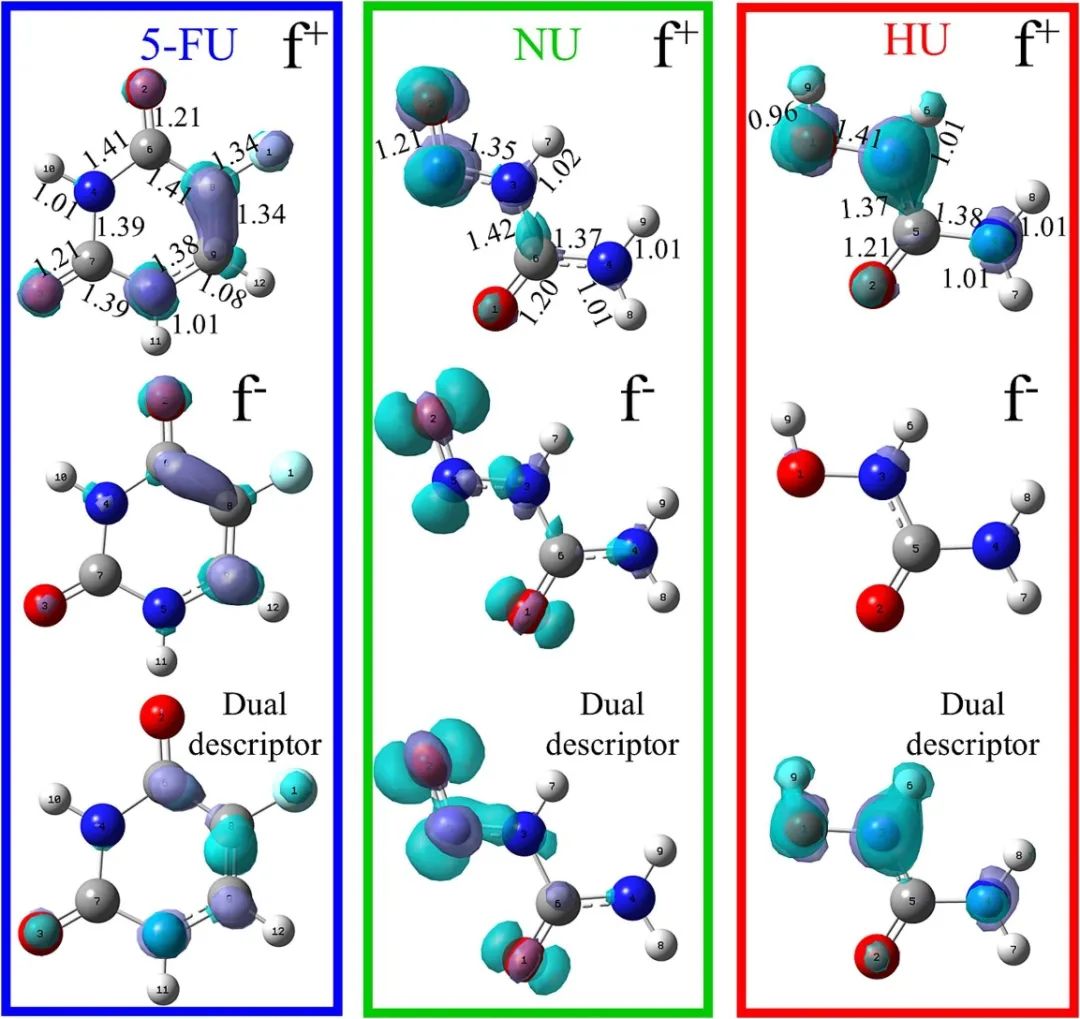
案例分析
绿色区域代表亲电攻击区,紫色区域代表亲核攻击区,直观展现分子反应位点分布倾向。结合表中凝聚福井函数值,能精准剖析不同环境(气相、水、DMSO等溶剂)下,各分子亲电、亲核攻击位点及活性顺序。
就5-FU而言,在气相环境中,通过凝聚福井函数计算与分析,明确亲电攻击位点为F1、O3、N5、C8。当处于不同溶剂环境,像水、DMSO、乙醇、丙酮等,溶剂极性会显著影响位点活性。在极性溶剂里,F1、O3、C8等原子的亲电攻击特性进一步增强,而N5原子作为亲电位点的反应活性有所减弱,亲电攻击位点顺序也随之改变;在低极性的氯仿环境中,顺序又有不同调整。
对于NU和HU抗癌分子,同样可依据福井函数及凝聚福井函数,明晰其在各环境下亲电、亲核攻击位点的活性变化规律。这种对不同环境下分子反应位点及活性的深入探究,助力科研人员透彻理解抗癌分子反应特性,为进一步探究它们与生物靶点的作用机制筑牢基础。
比如,知晓特定位点易受亲电或亲核攻击,就能针对性设计药物分子与靶点的相互作用方式,进而优化抗癌活性,为抗癌药物研发与改进提供关键理论依据,推动抗癌药物朝着更高效、更精准的方向发展。
7. 配位不饱和度
配位不饱和度的概念
配位不饱和度(Coordination Unsaturation)是配位化学领域中的一个核心概念,它主要用于描述配位化合物中金属中心周围的配位环境。在配位化合物中,金属原子或离子通过配位键与配体相结合,形成特定的空间结构。
配位不饱和度指的是金属中心周围的配位位置未被配体完全占据的程度,即金属中心的实际配位数小于其最大可能配位数的情况。最大可能配位数取决于金属原子的电子构型、空间几何因素以及配体的性质等。
配位不饱和度在配位化学中具有重要意义,它直接影响配位化合物的稳定性、反应活性和催化性能。具有较高配位不饱和度的配合物,由于金属中心存在未被占据的配位位置,使其具有更强的反应活性,更容易与外界的反应物分子发生配位作用,从而引发化学反应。在催化领域,配位不饱和度是影响催化剂活性的关键因素之一。
配位不饱和度与活性位点的联系
配位不饱和度对活性位点的电子结构和反应活性有着显著的影响。当配位不饱和度存在时,金属中心的电子云分布会发生改变。由于配位位置的空缺,金属中心的电子云不再呈对称分布,这会导致金属中心的电子密度和电子能级发生变化。
在一些过渡金属配合物中,配位不饱和度会使金属中心的d轨道电子云发生畸变,d轨道的能级分裂情况也会发生改变。这种电子结构的变化会直接影响金属中心与反应物分子之间的相互作用。反应物分子更容易接近具有配位不饱和度的金属中心,并与之形成配位键。
但如果配位不饱和度过高或过低,可能会导致反应中间体不稳定,或者反应物分子难以与活性位点发生有效作用,从而降低反应活性。因此,通过调控配位不饱和度,可以优化活性位点的电子结构和反应活性,提高材料在各种化学反应中的性能。