

理想电催化剂需具备超高本征活性、大量暴露活性位点、优异导电性、良好传质能力、出色电化学稳定性及低生产成本,而实现理想电催化剂构建需明确活性位点内部结构(物理结构、电子结构、电化学结构等)与多相电催化性能的构效关系,且电催化剂内部结构主要取决于活性位点的实际配位环境,该环境涵盖配位数、成键类型、空间位阻、键长与键角、活性中心密度等方面。
调控活性位点配位环境可优化电催化剂中活性物种的物理结构、电子构型及轨道杂化状态,进而精准调控电催化反应机制与效率,如优化中间体吸附/脱附能力以提升本征活性、抑制活性组分溶解以增强稳定性、丰富活性物种类型以提升催化剂功能通用性。
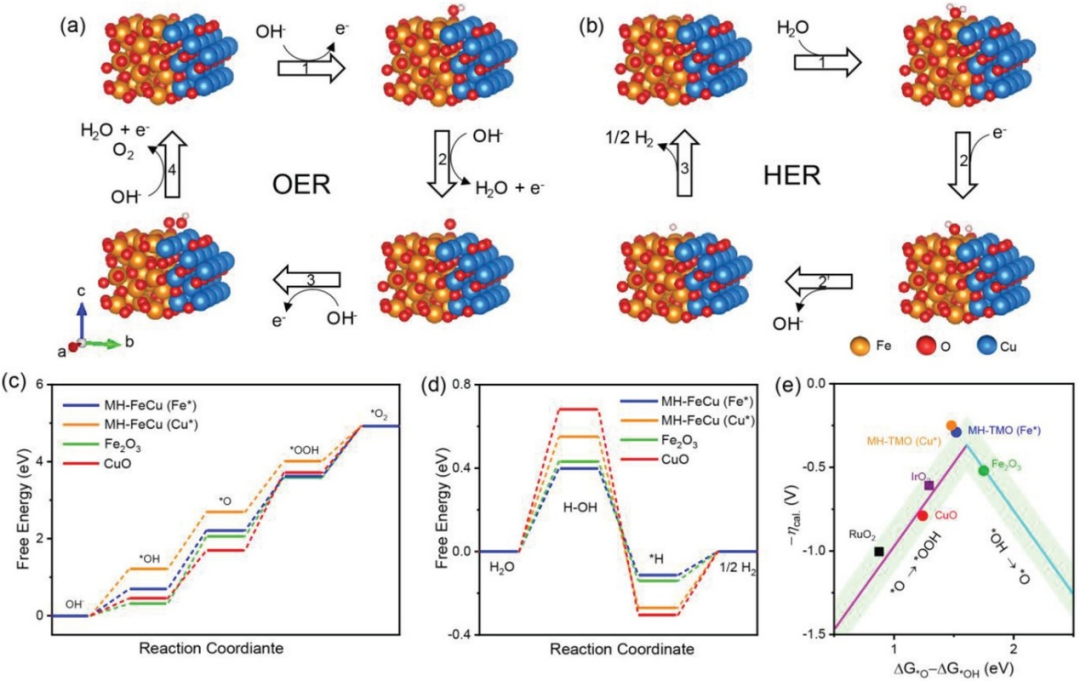

图1. DFT计算结果。(a,b)在MH-TMO(Fe位点)上,OER(a)与HER(b)反应中间体的吸附构型。(c,d)MH-TMO(Fe、Cu位点)、Fe₂O₃和CuO的OER(c)与HER(d)自由能图。(e)各催化剂上计算得到的负过电位(–ηcal)随描述符ΔGO*–ΔGOH*的变化关系。DOI: 10.1002/aenm.202200067
通常,活性位点的配位环境对其物理结构、电子构型及化学性质具有显著影响。此外,优化后的配位环境会进一步诱导电催化过程中活性位点的动态变化及其对中间体的吸附/脱附能力,使催化剂呈现出独特的电催化稳定性、活性与选择性。
配位键信息
活性位点的成键信息(键型、键长、键角等)是配位环境的基本要素。其中,键型对激活新活性物种并提升本征活性尤为关键。
如图2所示,研究人员设计了一种超薄(~1.6nm)PtRuRhCoNi高熵合金(PtRuRhCoNi-HEA)纳米线,在AOR与HER中均表现出超高质量活性(7.68A mg⁻¹ PtRuRh,11.99A mg⁻¹ PtRuRh)和稳定性。
密度泛函理论(DFT)计算表明,PtRuRhCoNi-HEA中金属位点丰富的键型诱导了成键轨道与反键轨道之间的强耦合,优化了金属活性中心的配位环境与电子构型,从而为AOR和HER提供了丰富且高活性的物种。
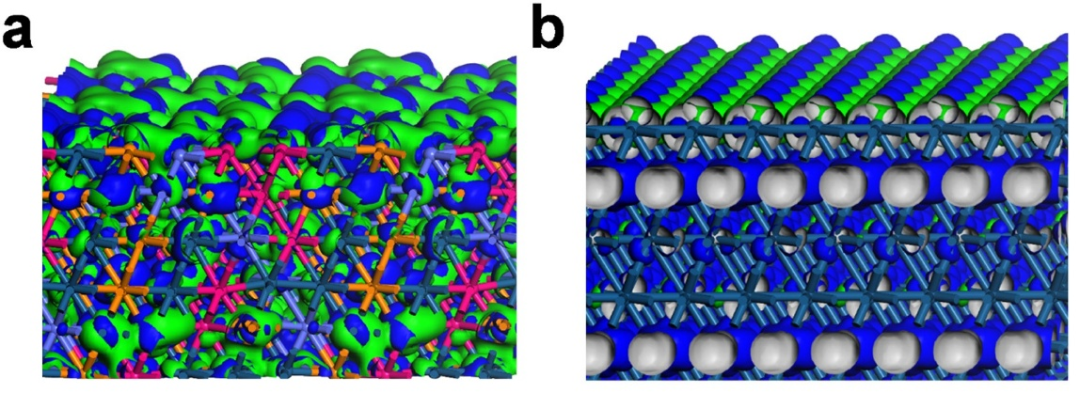
图2. (a)PtRuRhCoNi-HEA与(b)Pt在费米能级附近电子分布的三维等高图。
深绿色球=Pt,紫色球=Ru,橙色球=Rh,蓝色球=Co,粉色球=Ni。蓝色等值面=成键轨道,绿色等值面=反键轨道。DOI: 10.1016/j.apcatb.2022.121431
配位数
配位数作为判断催化剂配位环境的重要因素,在催化剂结构调控中发挥关键作用。通常,配位数较低的活性位点处于不饱和配位状态,与反应物接触时易通过电子轨道重叠结合反应物;这种强相互作用力有利于提高活性中心对反应物的吸附效率,但反应物的强吸附能力也可能导致活性位点中毒,不利于电催化性能提升。
如图3所示,研究人员指出,经Ar等离子体处理的IrO₂(CN=5.76)其Ir-O配位数显著低于经O₂等离子体处理的IrO₂(CN=6.52)及普通金红石相IrO₂;DFT结合原位X射线近边吸收精细结构(XANES)测试证实,低配位数的IrO₂催化剂更易被氧化,且Ir-O键长缩短至1.6 Å,有效降低了OER反应能垒。
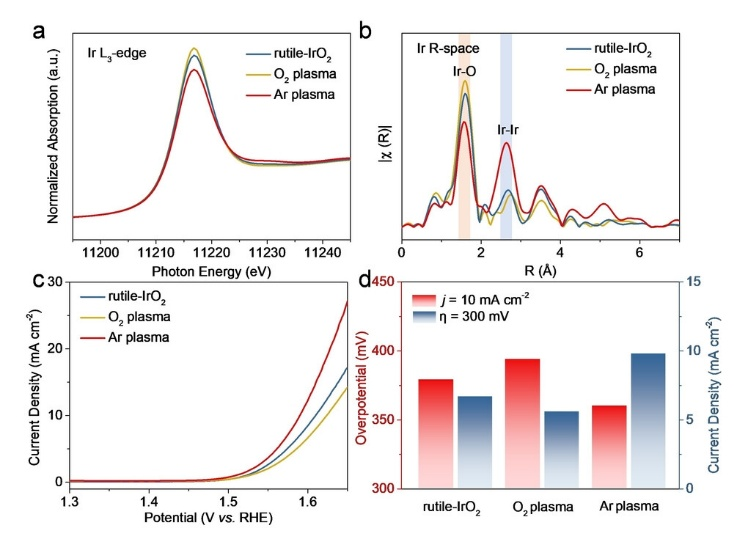
图3. (a)–(b)金红石型IrO₂及经O₂与Ar等离子体处理样品的Ir L₃边XANES谱(a)和R空间FT-EXAFS谱(b),用于确定各自的Ir–O配位数。(c)上述模型催化剂在5mV s⁻¹扫速下经iR补偿的LSV曲线。(d)各模型催化剂在10mA cm⁻²电流密度下的过电位η与300mV过电位下的质量活性对比。DOI: 10.1002/anie.202313954
空间位阻
空间位阻是配位环境组成的关键因素。具体而言,空间位阻主要指特定电催化体系中材料内活性位点的暴露程度,其对反应中间体的吸附、激活与解离具有重要调控作用,进而决定材料在催化功能上的选择性。
如图4所示,研究人员提出了一种空位诱导结构自调控策略,制备出高负载、高活性的单原子(SA)催化剂;DFT测试与晶体轨道分析表明,动态重构过程中金属空位迁移的能垒直接受单原子间距(即“单原子诱导空间位阻”)影响,且单原子间距最大的区域(即“空间位阻最小”)空位迁移能垒最低,有效提高了单原子负载量,进而优化了NO电还原性能。
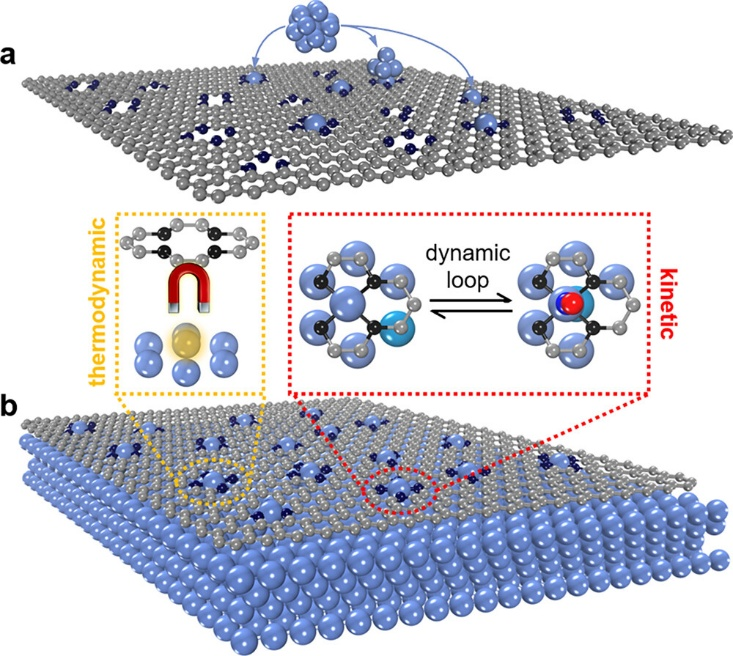
图4. (a)常规单原子催化剂(SACs)中缺陷对单原子(SAs)的捕获效率低。 (b)g/TM体系中,由于热力学自调节,双空位对SAs的捕获效率达100%,涉及吸附质辅助的可逆空位迁移动态循环。颜色标识:灰=C;黑=缺陷相关C;蓝=N;红=O;其他=过渡金属(TM)。DOI: 10.1021/jacs.3c08936
局部配位结构变形
随着对活性位点配位环境研究的深入,传统有序的配位结构被打破,局部配位结构变形(扭曲、无序、对称性等)逐渐进入研究视野。
如图5所示,研究人员利用M-Se(M=Ni、Fe)键长与M-O键长的不匹配性,在电化学表面重构(M-Se→M-OOH)过程中首次生成了具有高度无序与扭曲配位结构的M活性位点;
DFT测试证实,局部配位结构的扭曲与无序是改善局部电子构型、优化OER反应能垒的根本原因。通过调控局部配位结构变形构建稳定高效的活性位点,对推动电催化剂工业化应用具有重要指导意义。
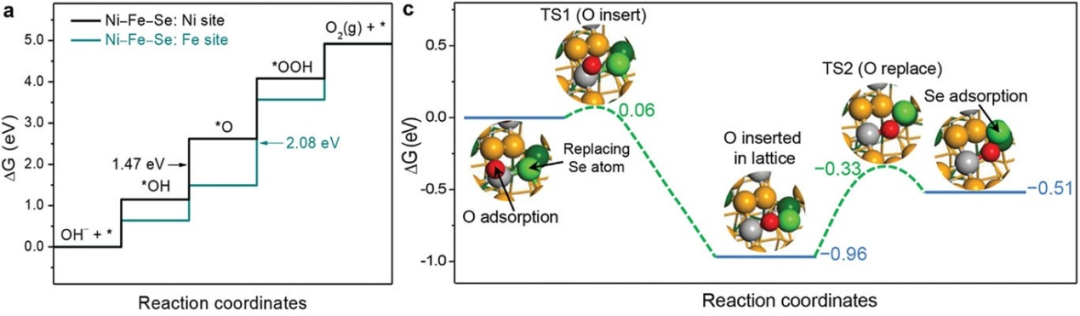
图5. (a)Ni–Fe–Se(210)表面模型上Ni位点与Fe位点的OER自由能图。(b)示意图:原始Ni–Fe–Se(210)(上)与重构后Ni–Fe–Se–OOH(下)表面Fe、Ni位点对O原子的吸附模型。绿色、灰、深黄、红、粉、白球分别代表Fe、Ni、Se、晶格O、吸附O与H原子。DOI: 10.1002/adma.202103004
活性位点密度
活性位点密度的调控作为配位环境的重要组成部分,往往容易被忽视。众所周知,传统M-N-C催化剂在ORR与OER领域具有较高本征活性,但金属活性位点密度低、传质阻力大等问题严重限制了其进一步发展。
如图6所示,研究人员利用小尺寸的CN⁻构建了高活性位点密度的Co(CN)₃微晶;X射线吸收光谱(XAS)证实,Co(CN)₃与传统Co-N-C材料具有相同的配位键,但Co(CN)₃中Co位点密度显著提高,为优化ORR活性奠定了基础。因此,从调控活性位点密度的视角优化配位环境具有重要意义。
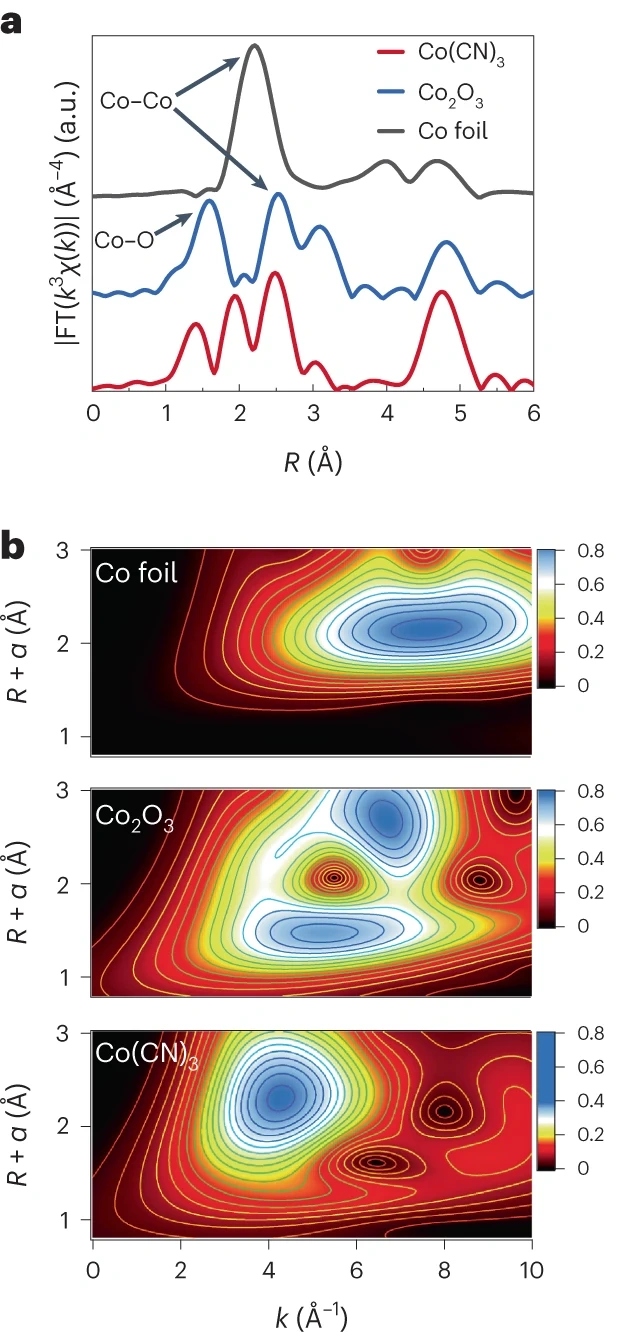
图6. (a)Co(CN)₃-Cub、Co₂O₃及钴箔的Co K边k³加权EXAFS傅里叶变换幅值;未做相移校正。(b)Co(CN)₃-Cub、Co₂O₃与钴箔的EXAFS小波变换图。DOI: 10.1038/s41929-023-01047-7
通过调控配位环境,可大幅优化催化过程中活性位点的结构及其对反应中间体的吸附/脱附能力,进而提升催化剂本征活性;同时,新形成的配位环境可能抑制活性位点溶解,提高电催化剂在实际应用中的耐久性;此外,配位环境的变化还可诱导活性物种富集与活性位点转移。
反应中间体吸附/脱附能力
作为制约电催化动力学的核心因素,反应中间体吸附/脱附能力需重点分析。配位环境可通过调控活性位点的d带中心位置、电子结构构型及轨道杂化状态,调节反应中间体的吸附/脱附能力,进而优化电催化剂本征活性。
如图7所示,研究人员将相邻的Fe-N₄与Ru-N₄位点锚定在氮掺杂碳骨架中,以调节Fe/N/C催化剂中Fe-N₄位点对ORR中间体的吸附强度;物理表征结合DFT计算表明,相邻原子分散的Ru-N位点降低了Fe位点的d带中心,减弱了Fe-O的结合亲和力及Fe-N₄位点对氧中间体(OH*)的吸附。
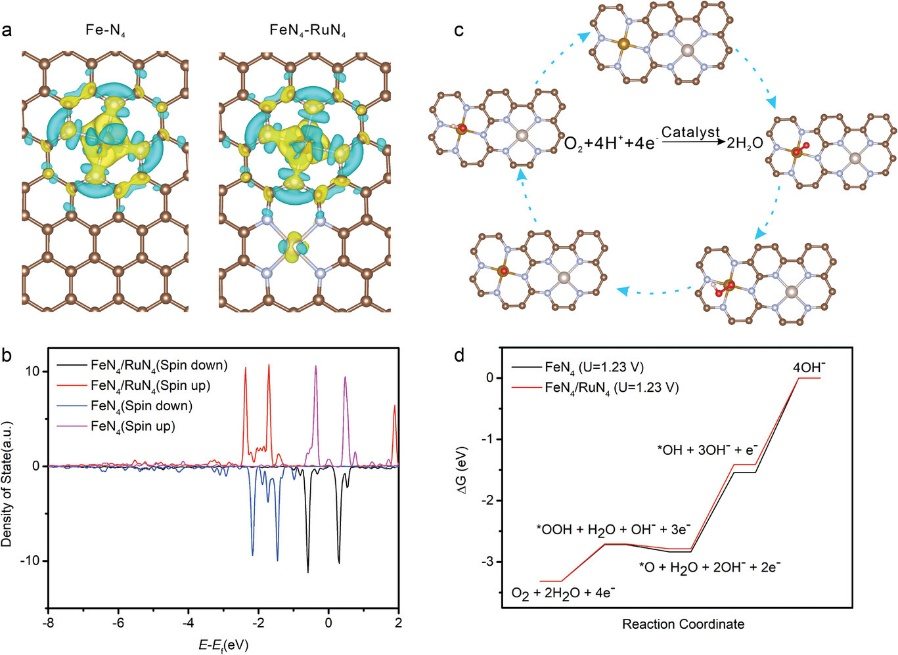
图7. (a)RuN₄与FeN₄/RuN₄的电荷密度差图。(b)FeN₄/RuN₄及FeN₄中Fe的d轨道投影态密度(PDOS)计算结果。(c)在FeN₄/RuN₄上提出的ORR反应机理示意图。(d)在1.23V下FeN₄/RuN₄与FeN₄的吉布斯自由能图。DOI: 10.1002/smll.202205283
活性位点稳定性
稳定性作为判断催化剂材料能否投入实际生产应用的重要指标,在催化剂设计过程中必须重点关注。许多催化剂在电催化过程中易出现活性位点溶解、失活与性能劣化等问题,严重阻碍其商业化进程。要从根本上提高电催化剂稳定性,掌握配位环境对稳定性的调控机制至关重要。
如图8所示,研究人员在Ce-Cu₂O材料中构建了独特的Ce⁴⁺ 4f-O₂p-Cu⁺ 3d结构;Ce⁴⁺ 4f-O₂的强相互作用抑制了晶格氧流失,促进Cu⁺活性位点稳定存在;且Ce⁴⁺ 4f-O₂p-Cu⁺ 3d结构中Ce⁴⁺4f轨道与O₂p轨道的非常规耦合,降低了Ce-Cu₂O对反应中间体的吸附能,增强了CO₂RR动力学。
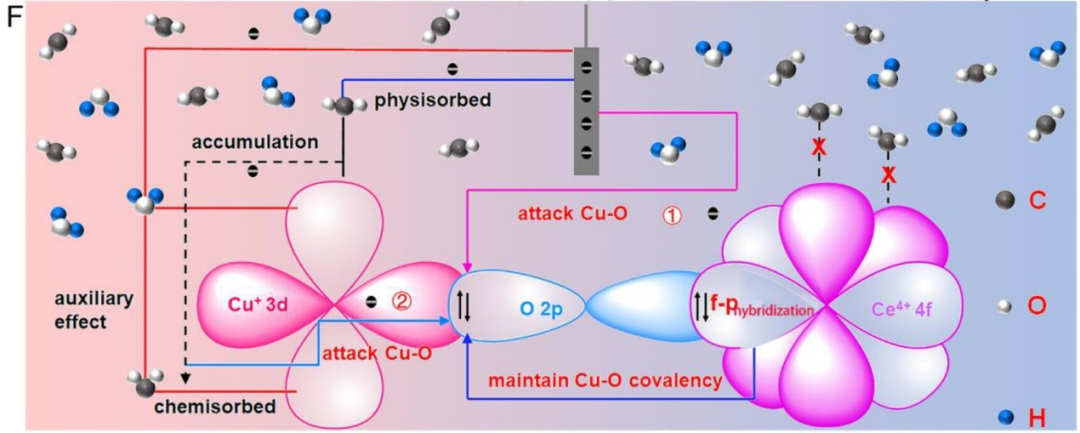
图8. (F)Cu⁺失活起源示意图:在CO₂RR过程中,Ce-Cu₂O中4f与2p高阶轨道耦合,维持Cu–O共价性,保护Cu⁺位点免受电子攻击,从而抑制Cu⁺失活。DOI: 10.1021/acsnano.3c03952
活性物种多样性
除活性相的本征活性与稳定性外,丰富电催化功能(即催化反应选择性)、实现催化剂在全pH电解质中的通用性,对电催化剂工业化应用至关重要。因此,通过改善活性位点配位环境丰富活性物种类型,对实现催化剂多功能性与通用性目标具有实际意义。
如图9所示,研究人员将Pt单原子(SA)与原子团簇(C)共同封装于氮掺杂多孔碳基质,制备出PtSA-PtC/NDPCM。Bader电荷分析显示,PtC位点的价电子范围(9.51–10.13)宽于PtSA,表明其具备更灵活的给电子能力;H原子可结合于不同类型Pt位点,实现中性/碱性条件下的卓越HER活性。
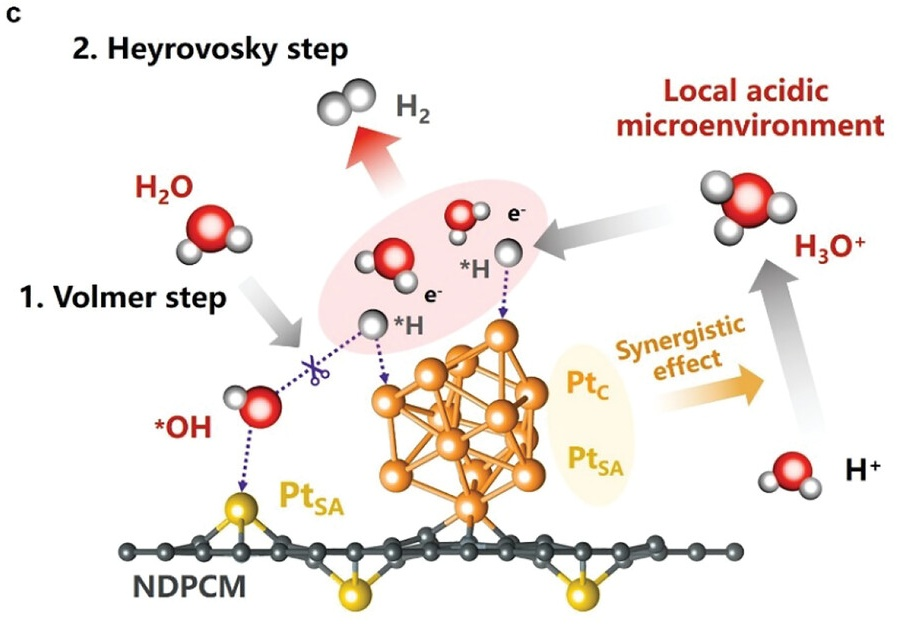
图9. (c)PtSA-PtC/NDPCM上HER过程机理示意简图。DOI: 10.1002/adfm.202304852
活性位点转化
随着活性中心配位环境的变化,活性位点可能发生相应的转移与转变,影响其电催化活性与稳定性。
如图10所示,研究人员通过在铁卟啉中调节FeXYᵢN3-i(X, Y=B, C, O, P, S;i=0,1)的配位环境,改变了Fe位点的自旋构型与电子分布,使HER、OER与ORR的活性位点从传统Fe中心转移至非金属(B、C、O、P、S)位点。这种“活性位点随配位环境迁移”的新观点,颠覆了“孤立金属原子始终为固定活性位”的传统认知。
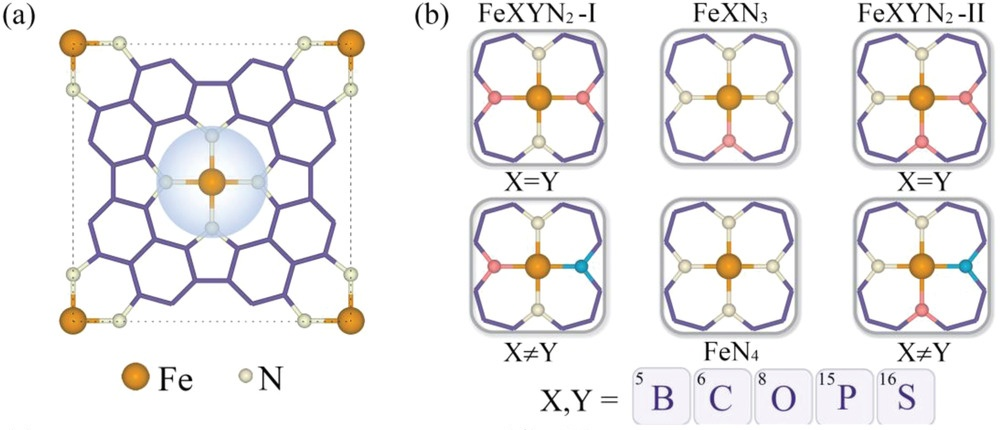
图10.(a)铁卟啉结构图,虚线圆圈标出FeN₄配位环境。(b)通过调控中心Fe原子第一配位壳层构建的FeXYᵢN3-i(i = 0, 1)活性中心示意图,包括单原子掺杂FeXN₃、同种双原子掺杂FeXYN₂-I(II)(X=Y)以及异种双原子掺杂FeXYN₂-I(II)(X≠Y);X、Y取自B、C、O、P、S。DOI: 10.1002/smll.202205111
#电催化#活性位点#配位环境#配位键#配位数#空间位阻#吸附#脱附#中间体#华算科技
