

说明:本文华算科技介绍了催化活性中心的定义、核心特征(电子结构、配位环境、空间构型)、研究三阶段及表征方法(静态、原位及多表征联用)。读者可系统学习到活性中心本质及研究演进,了解如何通过多种技术精准识别活性中心,为催化研究提供支撑。
提到催化活性中心,很多人会误以为“催化剂表面所有原子都能催化反应”,但在1925年Taylor提出的“活性中心概念”早已颠覆这一认知——催化活性中心是催化剂表面或内部具有特定化学环境(电子结构、配位构型、空间位阻)的局部区域,仅这些区域能与反应物分子发生特异性相互作用,降低反应活化能,加速反应进行。
催化剂的催化活性由其结构中的特定功能区域主导,这些区域相当于催化反应的核心场所,而其他表面区域则不直接参与反应过程。
以负载型Pt/C催化剂为例,其活性中心主要集中在Pt纳米颗粒表面的低配位“边角位”和“台阶位”,这些位点因其电子结构特性(如电子云密度高)能够有效吸附并活化O2分子。
相比之下,位于颗粒内部或表面高配位“平台位”的Pt原子,由于其结构和电子特性限制,催化活性显著较低。
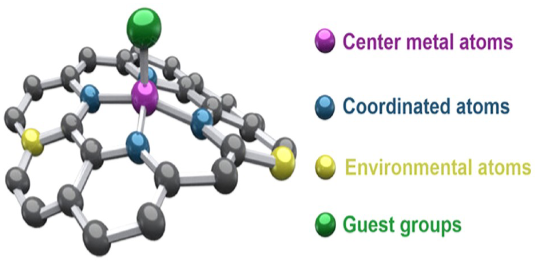

图1:催化剂示意图
判断一个位点是否为真正的活性中心,关键看其电子结构、配位环境、空间构型三大属性,这三者直接决定催化效率与选择性:
要包括活性中心原子的价态、电子云密度、d轨道能级分裂(过渡金属催化核心)。以过渡金属的“d空穴”为例——d空穴是d能带上有能级无电子的状态,既能接受反应物分子的电子,又能向反应物反馈电子,是氧化还原反应的“电子桥梁”。
比如Ni-Cu合金的案例极具代表性:纯Ni有0.6个d空穴,对乙烯加氢活性极高;当Cu含量增加到40%以上时,Cu的s电子会填满Ni的d空穴,合金的磁化率(反映d空穴数量)骤降,加氢反应速率常数也随之大幅下降,直接证明d空穴数量与催化活性的强关联。
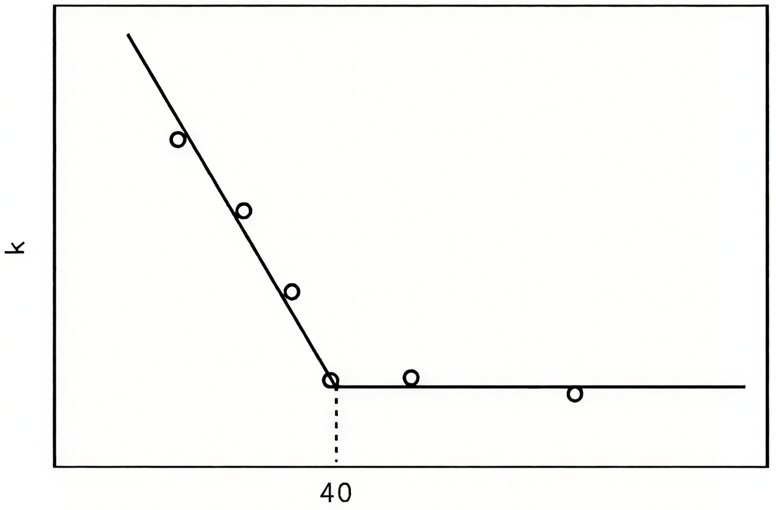
图2:合金中铜含量与反应活性的关系。link.cnki.net/urlid/61.1233.tq.20150616.1433.001
指活性中心原子周围的配位原子种类(如O、N、C)、配位数、键长。
比如CeO2表面的氧空位(Ovacancy),因周围Ce3+与Ce4+的配位失衡,形成未饱和电子态,能高效吸附O2并将其活化为超氧物种(O2⁻),成为CO氧化的关键活性中心;而TiO2表面的5配位Ti4+(体相Ti4+为6配位),因配位不完整,对H2O分子的吸附能力显著增强,光催化水解产氢活性比体相TiO2提升2倍以上。
包括活性中心的几何形状(凹陷位、凸起位)、相邻活性中心的间距,需与反应物分子的结构“匹配”才能高效活化。
比如工业氨合成反应中,Fe催化剂表面的“B5位”(由5个Fe原子构成的凹陷位),其空间构型与N2分子的线性结构高度契合,能通过多位点吸附削弱N≡N三键,活化能降低30%以上;
文献中通过原位表征发现,Fe(111)晶面的氨合成活性是Fe(110)晶面的430倍,核心原因就是Fe(111)晶面更易形成B5位活性中心。
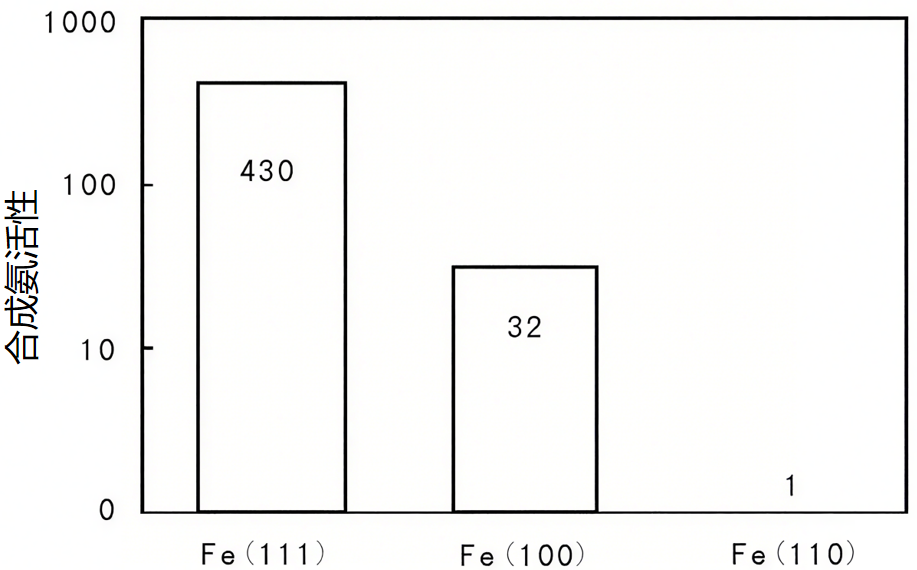
图3:在Fe不同晶面上氨合成活性
这一时期缺乏高分辨表征设备,研究者只能通过化学吸附、反应动力学曲线“间接猜结构”。
比如用程序升温脱附(TPD)测定活性中心数量——H2-TPD曲线中脱附峰的面积,对应金属催化剂表面可吸附H2的活性位点总数;
用红外光谱(IR)识别活性中心类型——CO在Pt表面吸附时,线式吸附峰(2000-2100 cm-1)对应低配位的边角位活性中心,桥式吸附峰(1900 cm-1)对应高配位的平台位。
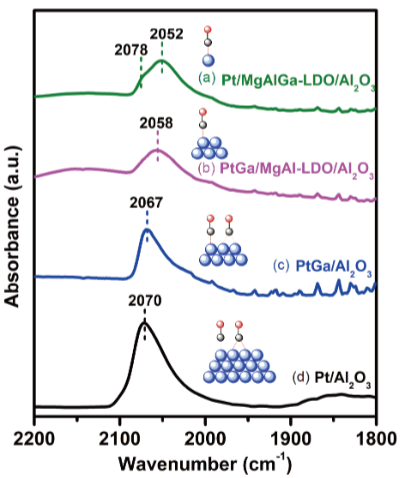
图4:CO的吸附
随着低能电子衍射(LEED)、X射线光电子能谱(XPS)、原位傅里叶变换红外光谱(in-situFTIR)等技术出现,研究者首次能在超高真空条件下观察单晶表面的活性中心。
比如德国Ertl团队(2007年诺贝尔化学奖)用LEED发现,Pt(111)晶面的“台阶位”原子(配位数7)比“平台位”原子(配位数9)更易活化CO,是CO氧化的活性中心;
CoMo/Al2O3加氢脱硫催化剂的研究更是经典——通过in-situ EXAFS(扩展X射线吸收精细结构)和in-situ FTIR,确认硫化后形成的“CoMoS相”(Co与Mo、S形成特定配位结构)是活性相,解决了该领域数十年的争议。
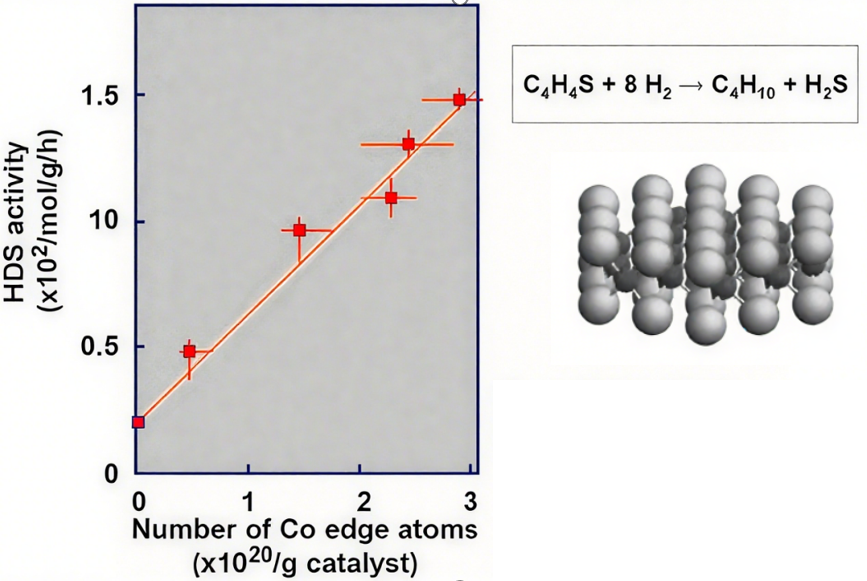
图5:CoMoS相和加氢脱硫活性关系图
纳米技术的兴起让活性中心研究从“静态观察”走向“动态调控”。
研究者可通过控制催化剂形貌,选择性暴露高活性晶面——比如优先暴露{110}晶面的Co3O4纳米棒,表面Co3+活性位点密度提升3倍,在-77℃有水汽存在时仍能实现CO完全转化。
包信和院士提出的“限域效应”更是颠覆性突破——碳纳米管管腔内的缺电子环境,能使Fe2O3的还原温度降低200℃,同时提高反应物局域浓度,让CO加氢制烯烃的选择性从40%提升到70%。
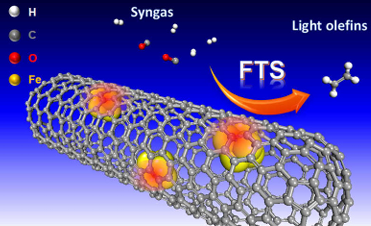
图6:碳纳米管管腔“限域效应”
1)化学吸附技术(TPD/TPR/TPO):测数量与强度
这是最经典的活性中心定量方法。程序升温还原(TPR)通过H₂消耗峰判断活性中心的还原难度——比如Fe2O3催化剂的TPR曲线中,500℃左右的峰对应Fe3+→Fe2+(活性前体),700℃左右的峰对应Fe2+→Fe0(活性中心),峰面积可量化不同价态Fe的含量;
程序升温脱附(TPD)则通过探针分子(如CO、NH3)的脱附峰位置,判断活性中心的吸附强度——峰温越高,吸附越强,比如Pt催化剂上CO的高温脱附峰(>300℃)对应边角位活性中心,低温峰()对应平台位(图5)。
2)X射线光电子能谱(XPS):分析化学态
XPS通过光电子结合能判断活性中心原子的价态——不同价态原子的核外电子云密度不同,结合能存在显著差异。
比如利用XPS分析Pt和Mo的价态,发现Mo掺杂使Pt的结合能红移,促进Pt从氧化态向金属态转变,形成电子富集的Pt0纳米颗粒。
Mo则主要以Mo6+存在,并向Pt传递电子。这些电子富集的Pt0颗粒暴露更多活性位点,显著提升催化剂的氢化性能。
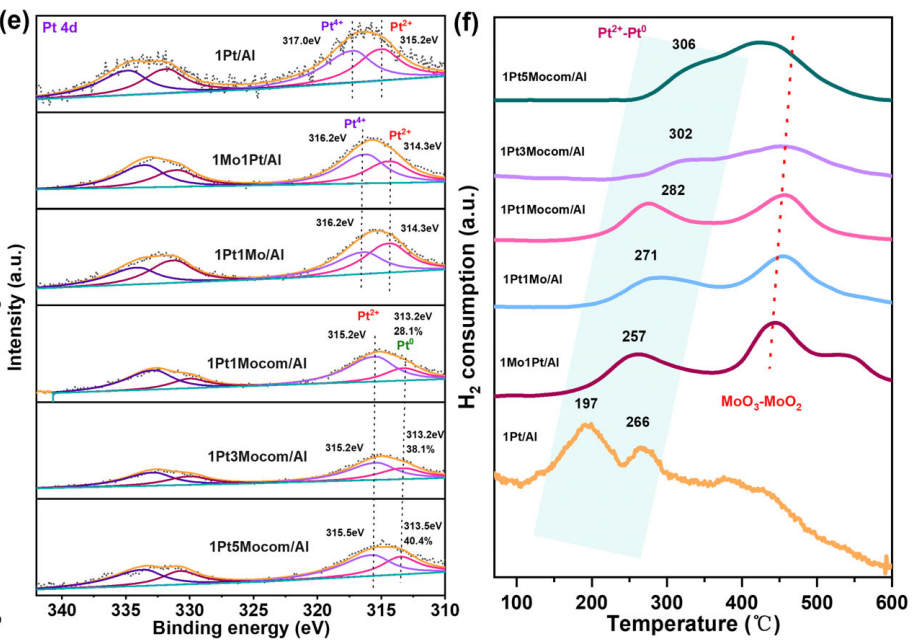
图7:XPS表征。DOI:10.1002/adfm.202505879
3)透射电子显微镜(TEM/HRTEM):看形貌与尺寸
高分辨TEM(HRTEM)可直接观察原子级结构,比如单原子Ni的引入显著增强了PtRu纳米合金与载体之间的电子金属-支撑相互作用(EMSI)。
在酸性和碱性介质中,PtRu-NiNC对HER和ORR均展现出卓越的催化活性与长期稳定性。具体而言,其HER过电位在酸性和碱性介质中分别仅为1.5 mV和26 mV,远低于PtRu-NC和商业Pt/C催化剂,证明PtRu-NiNC是主要催化中心。
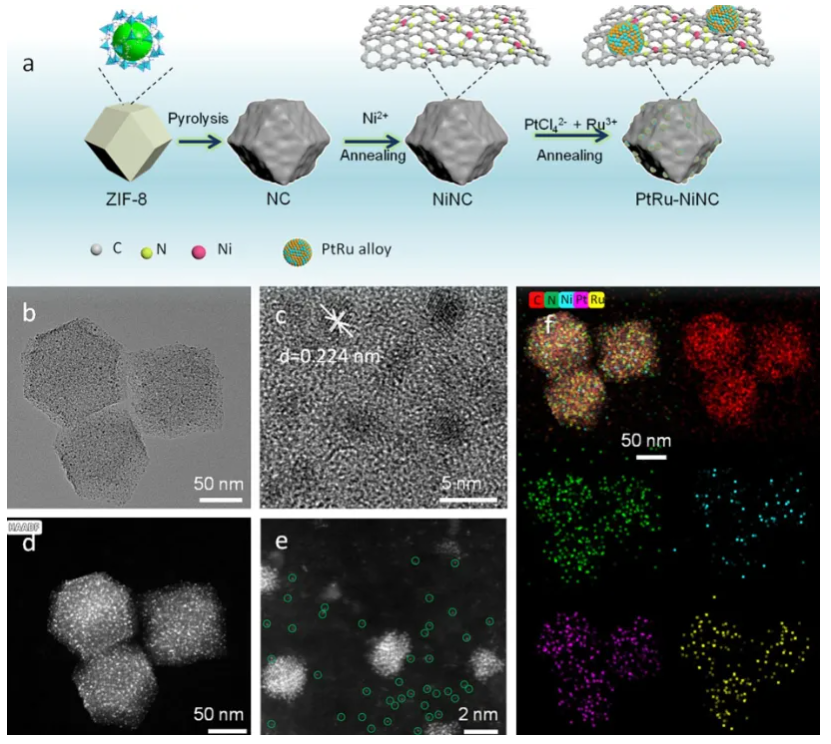
图8:展示了PtRu-NiNC催化剂的合成过程示意图以及相关表征结果。DOI:10.1016/j.jcis.2025.138271
1)原位傅里叶变换红外光谱(in-situ FTIR):追踪表面中间体
in-situ FTIR通过表面物种的红外吸收峰,实时观察活性中心与反应物的相互作用。
比如甲酸在Al2O3和ZnO上的分解反应,in-situ FTIR发现两者表面均有HCOO⁻吸附态(1572 cm-1),但Al2O3上的HCOO⁻峰强度不随反应变化(不参与反应),而ZnO上的HCOO⁻峰随反应逐渐消失(是反应中间体),直接区分了“旁观者”与“参与者”。
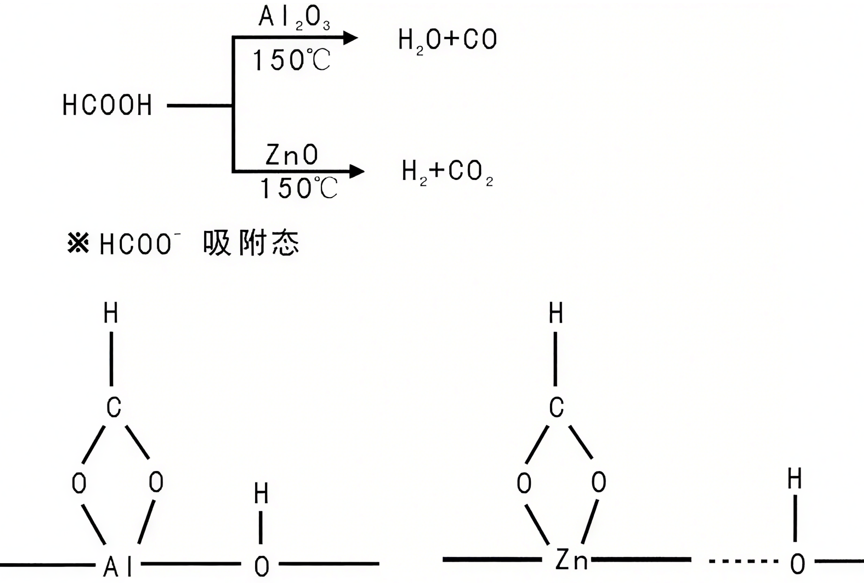
图9:HCOOH在Al2O3和ZnO上吸附物种
2)原位X射线吸收光谱(in-situXAS):同步获电子态与配位
XAS分为X射线吸收近边结构(XANES)与扩展X射线吸收精细结构(EXAFS),前者通过吸收边位置判断价态,后者通过傅里叶变换获取配位原子种类、配位数、键长。
对于HEA核心,Co和Ni的XANES光谱在高电位下变化不大,表明它们的溶解/氧化被抑制;而Fe和Cu的吸收边缘随电位增加向高能偏移,反映出它们在ORR过程中发生了氧化。
此外,N–Pt/HEA/C与Pt/HEA/C的白线强度差异分析表明,N掺杂增强了Co/Ni的稳定性,抑制了其溶解和氧化,但对Fe/Cu的影响较小。
这些结果表明,N掺杂引起的局部晶格畸变阻碍了核心中M原子的扩散,提高了N–Pt/HEA/C的耐久性,并确定了活性中心在ORR操作电位下的电子态和配位环境。
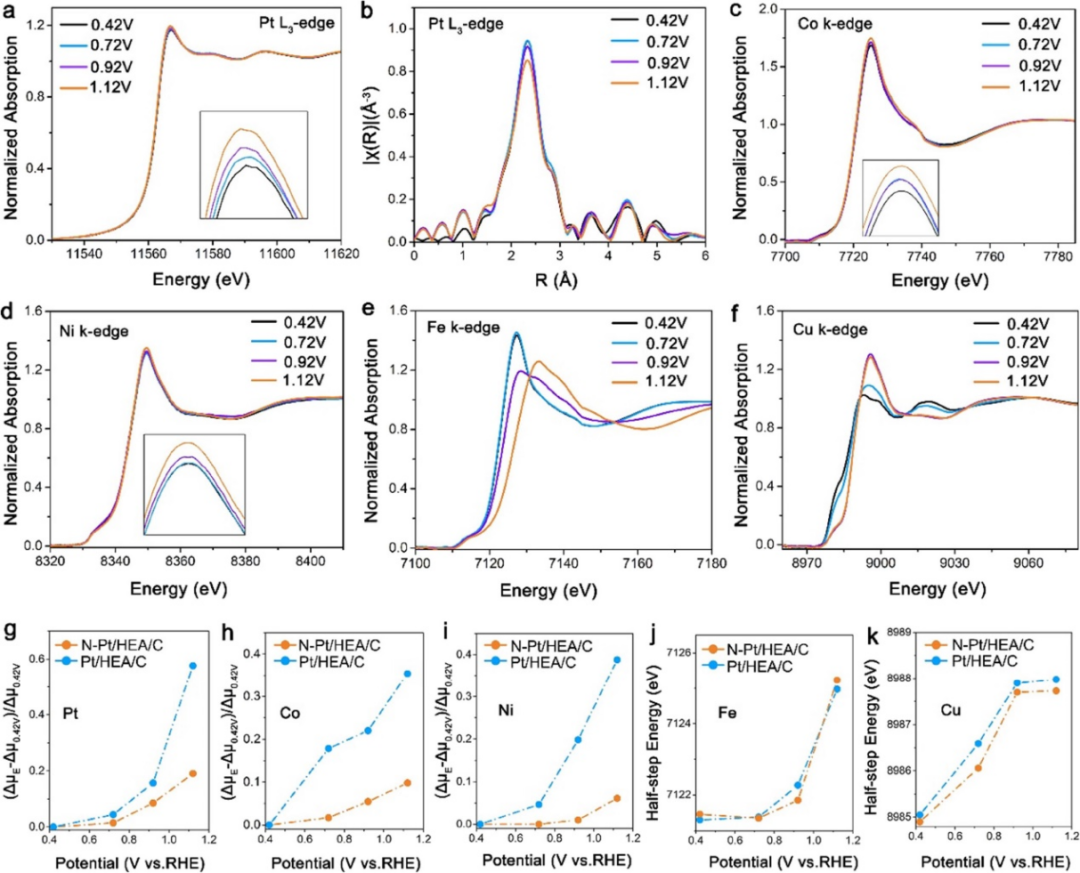
图10:N–Pt/HEA/C和Pt/HEA/C的原位XANES测量。10.1021/jacs.3c08177
3)原位透射电子显微镜(in-situ TEM/ETEM):看动态重构
在TEM样品室中构建微型反应池(可通气体、加热),实时拍摄活性中心的形貌变化。
比如Co3O4催化剂用于氧还原反应(OER)时,in-situ ETEM观察到反应前Co3O4为立方体结构,施加电压后表面逐渐形成无定形CoOOH层,且CoOOH厚度达5nm时OER活性最高,证明CoOOH是OER的活性中心。

图11:CO低温氧化的Co3O4纳米棒催化剂(左)与低温高效水汽变换反应的Au/CeO2催化剂(右)
单一表征技术无法全面揭示活性中心本质——XPS能测价态但看不到配位环境,TEM能看形貌但分不清活性位点与惰性位点,只有让不同表征互补验证,才能得到可靠结论。
案例1:双分子探针+in-situ FTIR+TPD,区分双金属活性中心
研究硫化态CoMo/Al₂O₃加氢脱硫催化剂时,单独用CO吸附IR无法区分Co和Mo中心(两者CO吸附峰重叠)。
辛勤团队提出“双分子探针法”:让CO与NO共吸附,通过in-situ FTIR观察到——CO仅吸附在Co中心(2020 cm-1),NO仅吸附在Mo中心(1850 cm-1),成功区分两种活性中心;再结合CO-TPD:Co中心的CO脱附峰在250℃(吸附弱),Mo中心在350℃(吸附强),证明Co能将吸附的H2转移到Mo活化的S原子上,解释了Co对Mo的助催化作用。
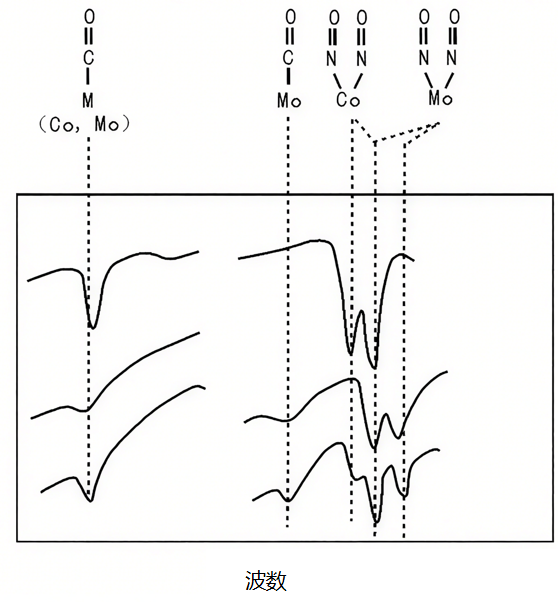
图12:CO和NO共吸附在硫化态Co-Mo/Al2O3的FT-IR
案例2:XRD+TEM+in-situXAS,锁定PtRu合金的活性中心
研究PtRu/C甲醇氧化催化剂时,XRD发现Pt的晶胞参数从3.916 Å缩小到3.883 Å,证明Ru进入Pt晶格形成合金;TEM观察到合金粒径约3 nm,分散均匀;
in-situ XAS显示Pt-Ru键配位数为4,键长2.7 Å,XANES确认Pt的d带中心下移——三者共同证明:PtRu合金通过“电子效应”(d带中心下移减弱CO吸附)和“几何效应”(Pt-Ru键缩短促进甲醇脱氢),使甲醇氧化活性比纯Pt提升5倍。
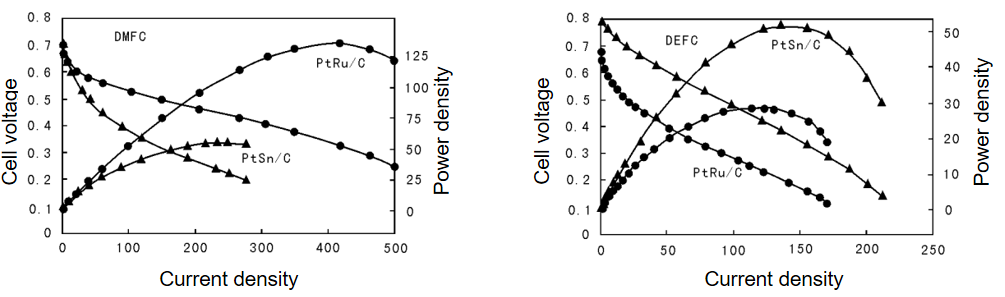
图13:Pt-Sn/C,Pt-Ru/C电催化活性比较
本文围绕了催化活性中心展开,先明确其定义为催化剂表面/内部具特定化学环境的局部区域,阐述电子结构、配位环境、空间构型三大核心特征,梳理经典、表面科学、纳米科学三研究阶段,还详解静态(TPD/TPR、XPS等)、原位(in-situ FTIR、XAS等)表征法及多表征联用案例。
未来,这些内容可助力研究者更精准识别活性中心,推动催化领域从经验设计迈向理性设计,为高效催化剂研发提供有力支撑。