

说明:在电催化领域,活性位点是实现高效电化学反应的关键所在。它不仅是电催化反应发生的微观场所,更是决定电催化性能的核心因素。本文华算科技将深入探讨电催化领域中活性位点的定义、特性、研究方法及其在实际应用中的重要性,帮助读者全面理解这一关键概念。
什么是活性位点
在电催化过程中,活性位点是指催化剂表面或内部能够与反应物分子发生有效相互作用,并促进电化学反应进行的特定区域。这些位点通常具有独特的电子结构和几何构型,能够显著降低反应的活化能,从而提高反应速率和选择性。
电催化反应涉及电子的转移过程,活性位点在这一过程中起到了“桥梁”的作用。它能够吸附反应物分子,稳定反应中间体,并促进电子的传递,最终生成目标产物。在氢气生成反应(Hydrogen Evolution Reaction,HER)中,活性位点能够吸附质子(H+),并通过电子转移将其还原为氢气(H2)。
如图1,可以得出结论,多孔NiW双金属合金电催化剂与单金属对应物相比,显示出增强的反应动力学和HER性能,这归因于W和Ni双重活性位点之间的协同效应,加速了H2O的分解并增强了H2的产生。。
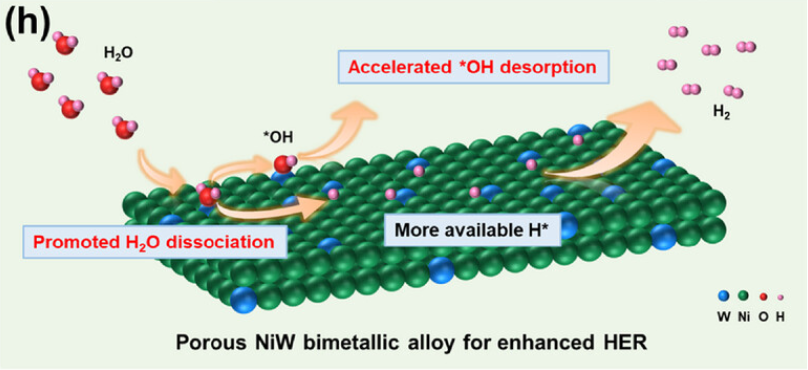

图1:提出的用于HER的NiW双金属合金反应机理。DOI:10.1002/adma.202503742
电催化活性位点的特性
电子结构特性
活性位点的电子结构是决定其催化性能的关键因素之一。活性位点的电子云密度、能带结构和电荷分布等特性直接影响其与反应物分子的相互作用强度和方式。
如图2在金属催化剂中,金属原子的d带中心位置对活性位点的电子结构起着决定性作用。d带中心靠近费米能级时,活性位点具有较强的吸附能力,但可能导致反应物过度吸附,从而降低反应速率;而d带中心远离费米能级时,吸附能力较弱,反应速率也会受到影响。因此,优化活性位点的电子结构是提高电催化性能的重要途径。
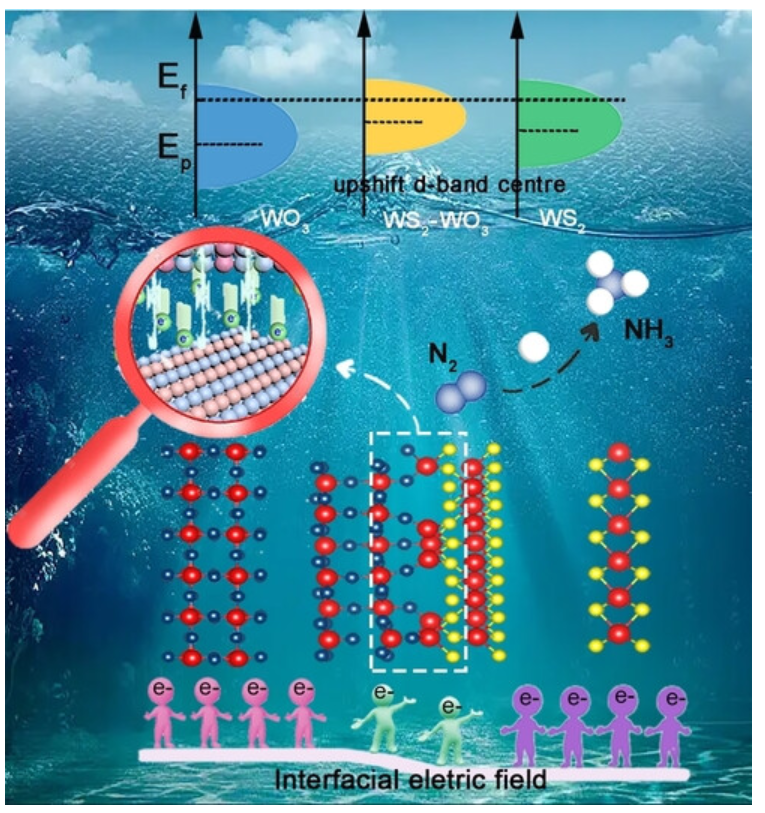
图2:WS2-WO3中的强界面电场通过提升W的d带中心,增强对中间体的吸附。DOI:10.1002/anie.202303794
几何结构特性
活性位点的几何结构同样至关重要。其形状、尺寸和表面原子排列方式等几何特性决定了反应物分子在其上的吸附位点和吸附方式。在纳米催化剂中,纳米颗粒的形状(如纳米线、球形、立方体等)和尺寸(亚纳米、纳米尺寸)对活性位点的暴露情况和反应物的扩散路径有显著影响。
如图3表明,当尺寸减小到一定程度时,通过共组装合成的高度有序排列的亚纳米线具有高比表面积,通常具有更多的活性位点,从而表现出更高的电催化活性。
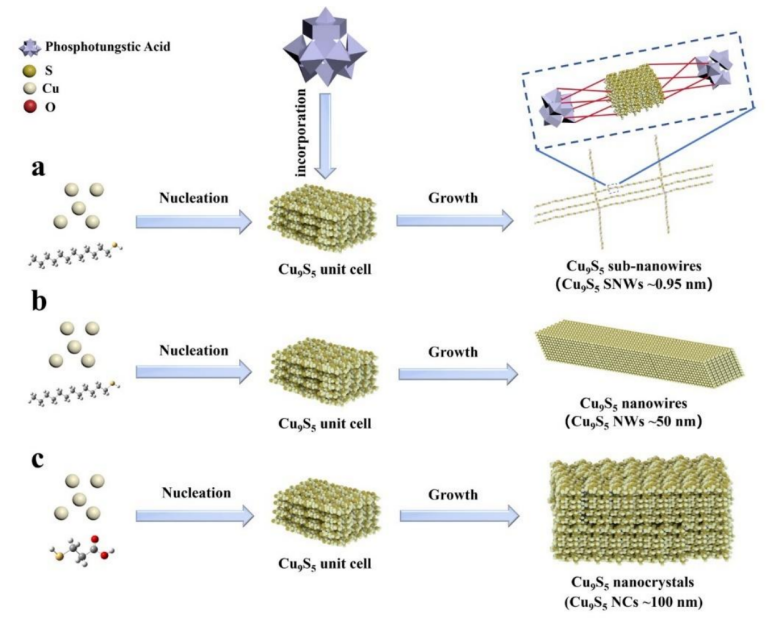
图3:不同尺寸Cu9S5 的制备示意图。DOI:10.1002/aenm.202403354
稳定性特性
在电催化反应中,活性位点的稳定性也是影响催化性能的重要因素。活性位点需要在反应过程中保持结构和功能的稳定性,以确保持续高效的催化反应。在酸性或碱性电解质中,活性位点可能会受到腐蚀或溶解,从而导致催化剂失活。
因此,提高活性位点的化学稳定性和电化学稳定性是电催化研究中的一个重要方向。通过合金化、表面修饰或构建复合结构等方法,可以增强活性位点的稳定性,延长催化剂的使用寿命。
如图4除了催化效率的提高外,还研究了引入O对稳定性的效果。在电流密度为20 mA cm-2的100小时长期测试中,SNDP-Pd@HEAA表现出良好的稳定性,其过电位仅下降约17.3 mV,远小于As-SNDP-Pd@HEAA的下降,证明催化剂的改性能够提高活性位点的稳定性。
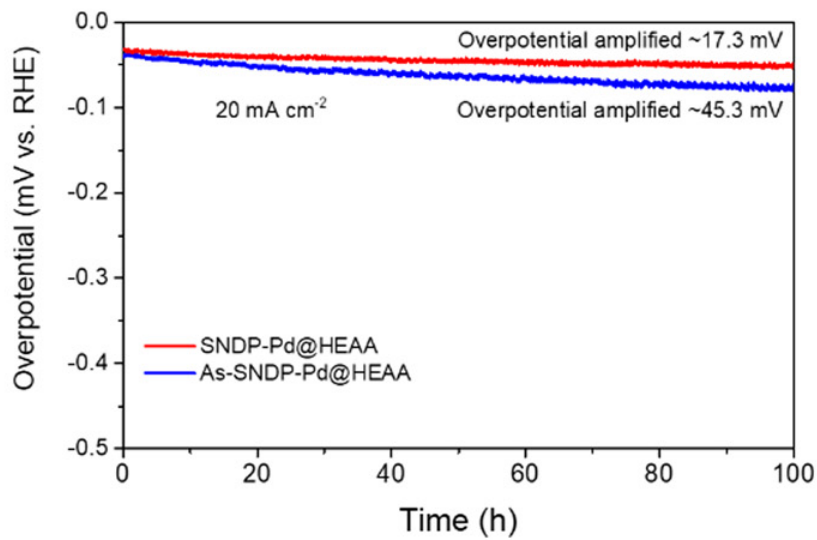
图4:SNDPPd@HEAA和As-SNDP-Pd@HEAA的稳定性测试。DOI: 10.1039/d4ee03150d
电催化活性位点的研究方法
实验表征方法
电化学测试技术
循环伏安法(Cyclic Voltammetry,CV):通过在电极和电解质之间施加周期性变化的电位,测量电流响应,可以评估活性位点的电化学活性。CV曲线的形状和面积反映了活性位点的数量和反应速率。如研究氢气生成反应时,CV曲线中的氢气析出峰面积越大,说明活性位点越多,催化活性越高。
电化学阻抗谱(Electrochemical Impedance Spectroscopy,EIS):通过施加一个小幅度的交流信号,测量电极系统的阻抗响应,可以分析活性位点的电子传递过程和电荷转移阻抗。EIS能够提供关于活性位点的电荷转移动力学信息,帮助优化催化剂设计。
表面分析技术
X射线光电子能谱(XPS):用于分析活性位点表面的元素组成和化学状态。XPS可以检测表面原子的价态和化学环境,从而推断活性位点的电子结构。在研究过渡金属催化剂时,XPS可以确定金属元素的氧化态,帮助理解其催化活性。
如图5,XPS分析表明SNDPPd@HEAA中Pd主要以Pd0存在,且因周围无定形结构有轻微能量偏移。引入氧后,改变Pd 3d信号,说明氧可调节其d带电子结构。O的XPS峰反卷积显示其与多种原子复杂配位,且O更倾向于与HEAA元素反应,而非氧化Pd,这表明HEAA可保护Pd,使O在非晶区富集。
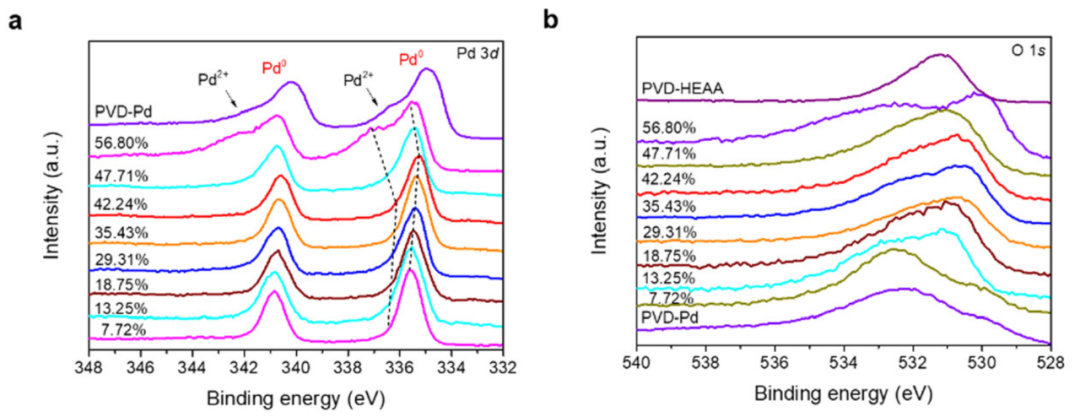
图5:SNDP-Pd@HEAA的XPS精细谱。DOI: 10.1039/d4ee03150d
原位光谱技术
原位拉曼光谱(In-situ Raman):原位拉曼光谱可以提供反应过程中活性位点的结构信息,可以研究活性位点与反应物之间的相互作用。
如图6通过置换反应在铜箔上原位生长了Bi@Bi2O3纳米树枝状催化剂,其在反应中重构为Bi纳米花,最大化暴露活性位点,增强CO2吸附和活化。
在-0.9 V(相对于RHE)的电位下,HCOOH的法拉第效率高达92.3%。原位拉曼光谱显示,在反应过程中,1469 cm-1处出现*OCHO中间体的拉曼峰(甲酸的关键前体),1130 cm-1处出现*COOH中间体的拉曼峰(CO生成的关键前体)。
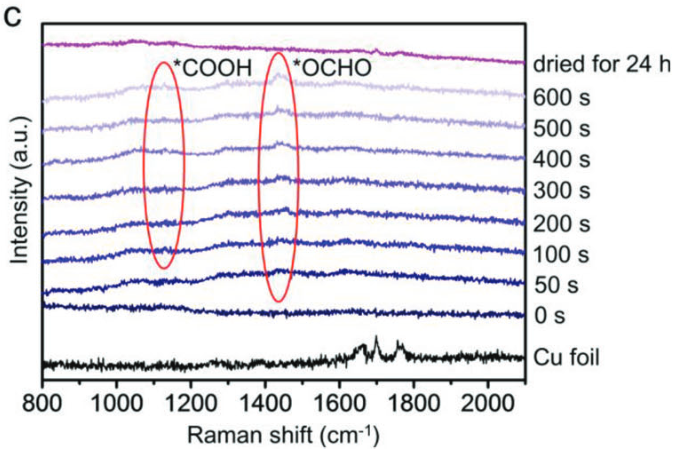
图6:在Bi-NFs表面于–0.9V对RHE记录的时间依赖性原位拉曼光谱。10.1002/adfm.202301984
理论计算方法
密度泛函理论(Density Functional Theory,DFT)
DFT是一种基于量子力学的计算方法,用于研究活性位点的电子结构和反应机理。通过计算活性位点的电子密度分布和能带结构,DFT可以预测活性位点的吸附能力和反应活性。例如,在研究金属催化剂时,DFT可以计算金属原子与反应物分子之间的吸附能,从而评估活性位点的催化性能。
H的吉布斯自由能(ΔGH*)是衡量HER性能的关键参数,理想的值接近零,以确保催化位点与H之间的相互作用适中,既能可逆吸附又能顺利解吸。
如图7利用密度泛函理论计算了SNDP-Pd@HEAA的ΔGH*,研究了H质子在晶体、非晶以及晶体/非晶界面模型催化剂表面Pd位点吸附后的ΔGH*和原子构型。结果表明,晶体/非晶态界面处的Pd位点比晶体和非晶态界面处的Pd位点具有更有利的ΔGH*值。
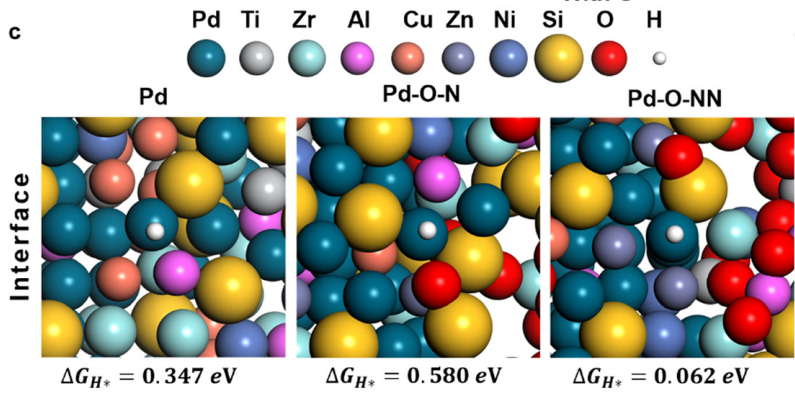
图7:密度泛函理论(DFT)模拟SNDP-Pd@HEAA在碱性条件下催化析氢反应。DOI:10.1039/d4ee03150d
总结与展望
活性位点在电催化中极为关键,它影响反应的活性、选择性、催化剂的稳定性和寿命。通过研究其电子结构、几何结构和稳定性,并结合先进实验与理论计算,研究人员可设计高性能电催化剂,推动电催化技术在能源转化和环保领域的应用。
未来,随着纳米技术、材料科学和计算科学的发展,活性位点研究将更深入。例如,构建多尺度模型并结合机器学习和人工智能,可加速新型催化剂的设计和优化。活性位点研究既是科学问题,也是重大的技术挑战。最后希望本文能帮助读者全面了解其重要性,吸引更多科研人员投身这一充满挑战与机遇的领域。