

什么是催化火山图


在催化反应的理论研究领域中,催化火山图是一种用于描述催化剂活性或选择性与催化剂表面吸附能(或其他描述符)之间关系的重要工具。
其得名源于图像呈现出类似火山形状的曲线,在曲线的中间位置存在一个峰值,向两侧活性或选择性逐渐降低 ,这一特殊的曲线形态蕴含着催化反应中深刻的内在规律。
火山图的横坐标通常代表一个描述符变量,这个变量与催化剂的电子结构或几何结构密切相关,能够反映催化剂活性中心的性质变化。常见的描述符变量包括金属原子的d带中心位置、催化剂的电子密度、活性位点的某种结构参数、反应物在催化剂表面的吸附自由能(如OER中的吸附自由能 ΔG*OH等) 。
纵坐标则一般代表催化剂的活性、选择性或转化率等性能指标。这些指标用于衡量催化剂在特定催化反应中的表现,数值越高表示催化剂的性能越好。
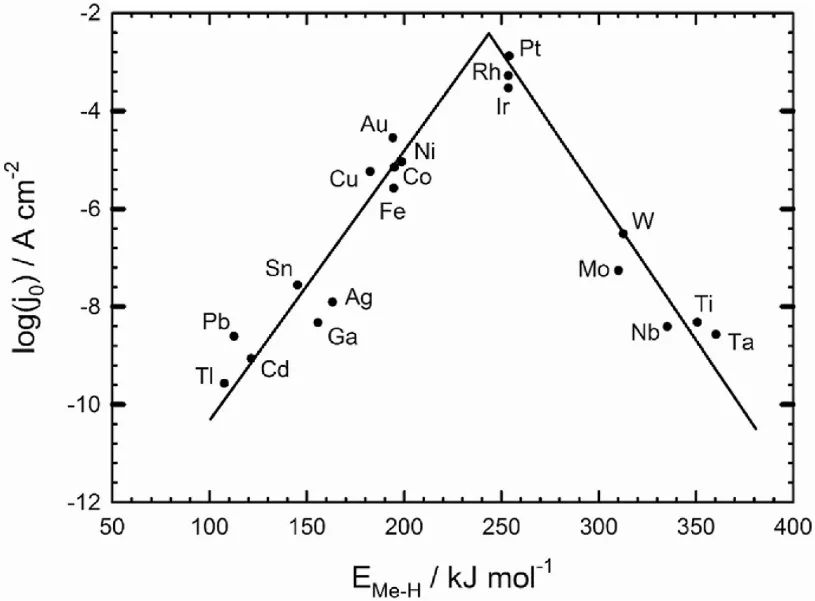
从本质上来说,催化火山图建立在对催化反应过程的深入理解之上。催化反应的发生依赖于反应物在催化剂表面的吸附和脱附过程,以及吸附物种之间的反应转化。
而催化剂表面对反应物的吸附能是影响这些过程的关键因素。如果催化剂对反应物的吸附过强,反应物会牢固地吸附在催化剂表面,难以发生后续的反应转化,并且阻碍了产物的脱附,导致催化剂活性降低。
反之,如果吸附过弱,反应物难以在催化剂表面富集,反应难以有效进行,同样会使催化活性不理想。只有当吸附能处于一个合适的中间范围时,催化反应才能以最高的效率进行,从而对应着火山图曲线的峰值。
火山图的特点
对称性与非对称性
催化火山图的形状在不同的催化反应体系中呈现出多样性,其中对称性和非对称性是其重要特征之一。理想情况下,火山图可能呈现出对称的形状,即曲线以峰值为中心,左右两侧的下降趋势基本相同。
这种对称性反映了在催化反应中,吸附能对催化活性的影响具有某种程度的均衡性。也就是说,当吸附能偏离最优值时,无论是吸附过强还是过弱,对催化活性的负面影响程度相似。
例如,在一些简单的小分子吸附和反应体系中,如氢气在金属表面的解离吸附和后续反应,如果反应过程相对单一,且涉及的中间物种较少,可能会观察到较为对称的火山图。
然而,在实际的催化反应体系中,更多出现的是非对称的火山图。这种非对称性源于催化反应过程的复杂性。不同的吸附物种在催化剂表面的吸附和反应行为存在差异,反应路径可能存在多个分支,以及催化剂与反应物、中间物种和产物之间的相互作用具有多样性。
例如,在多步反应过程中,某些关键中间物种的吸附能对反应速率的影响更为显著,导致当吸附能偏离最优值时,向某一方向的变化会使催化活性下降得更快。此外,催化剂表面的重构、电子效应在不同方向上的差异等因素,也会打破火山图的对称性。
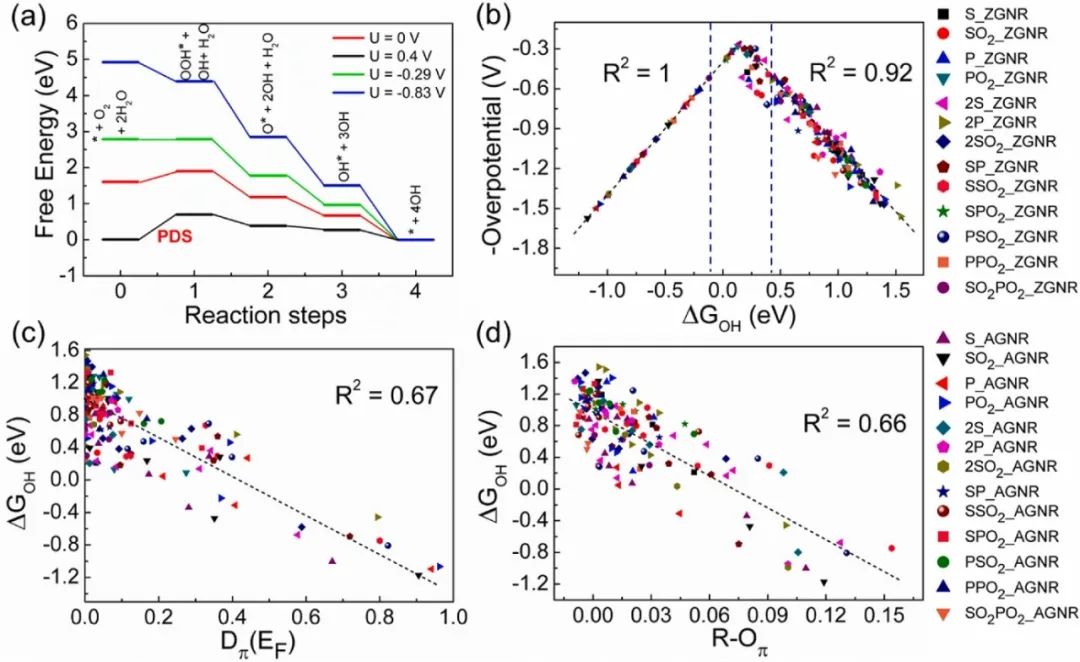
极值点的物理意义
火山图的极值点(即峰值)具有重要的物理意义,它对应着催化反应中最佳的催化剂性能。
在极值点处,催化剂表面的吸附能处于一个恰到好处的状态,使得反应物能够有效地吸附在催化剂表面,发生必要的化学反应,同时产物也能够顺利脱附,从而实现最高的催化活性或选择性。
从电子结构和化学吸附的角度来看,在极值点所对应的催化剂体系中,催化剂与反应物之间的电子转移和轨道相互作用达到了一种平衡。
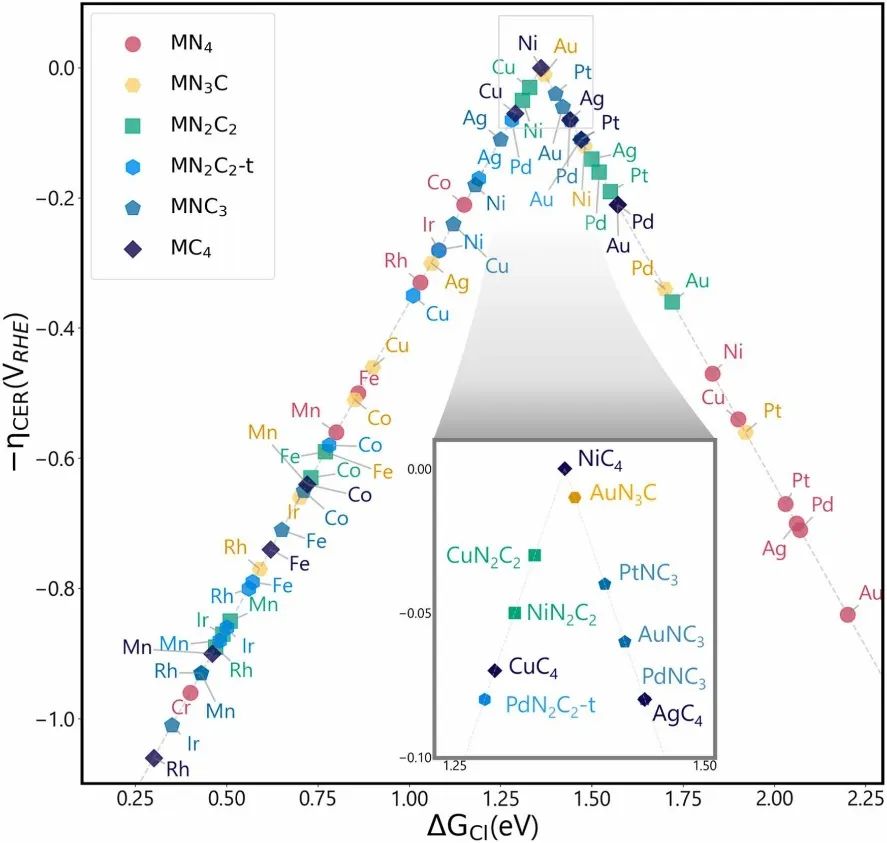
数据离散性与拟合效果
在通过理论计算构建催化火山图时,数据的离散性是不可避免的问题。由于催化反应体系的复杂性,以及理论计算方法本身存在的一定近似性,计算得到的数据点往往不会完美地分布在一条光滑的曲线上。
不同的计算模型、基组选择、计算参数设置等因素,都会导致计算结果存在一定的误差和波动。此外,催化反应过程中存在的多种相互作用和动态变化,也使得实际的催化性能难以用简单的数学关系精确描述。
为了更好地呈现催化活性与描述符之间的关系,通常需要对计算得到的数据进行拟合。常用的拟合方法包括线性拟合、多项式拟合、非线性拟合等。拟合效果的好坏直接影响到火山图对催化反应规律的准确表达。
良好的拟合能够有效地平滑数据点的离散性,突出整体的变化趋势,清晰地展现出火山图的特征。然而,如果数据离散度过大,或者选择的拟合方法不合适,可能会导致拟合曲线无法准确反映真实的催化性能变化规律,甚至产生误导性的结论。
因此,在构建催化火山图时,合理选择拟合方法,并对拟合结果进行充分的验证和分析至关重要。
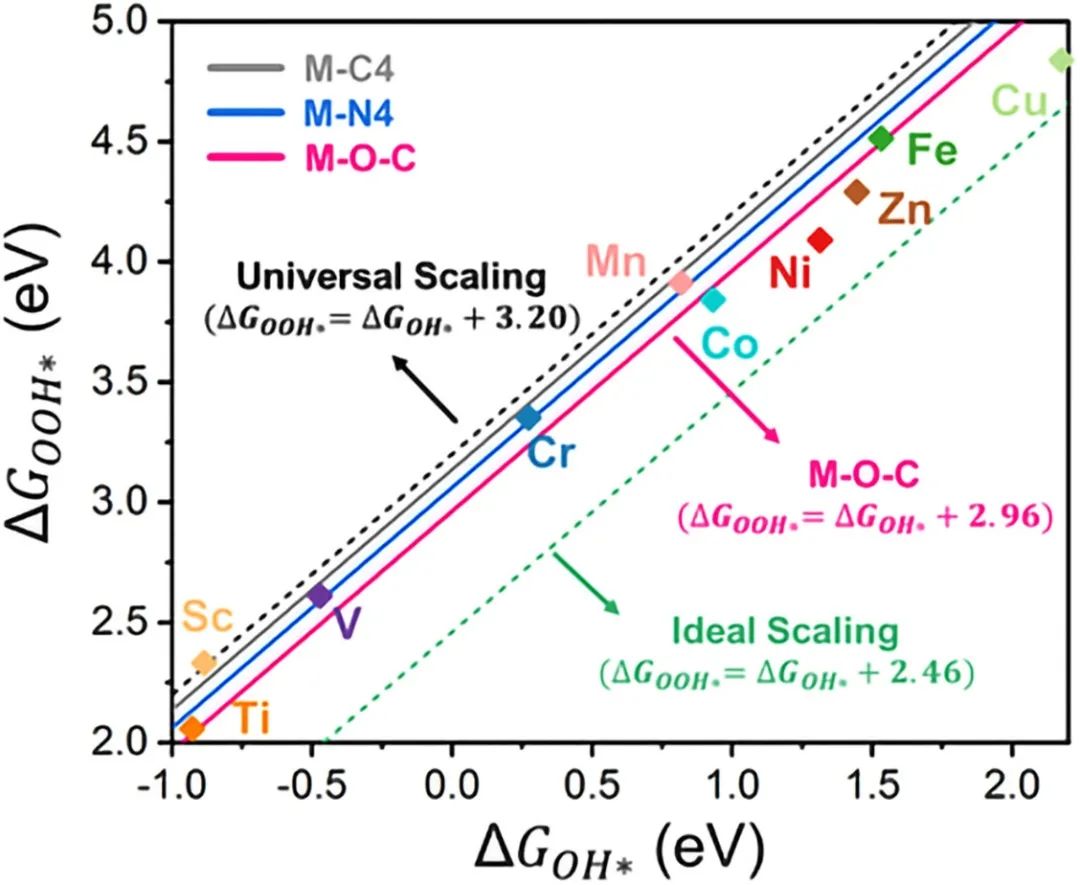
火山图案例分析
案例1
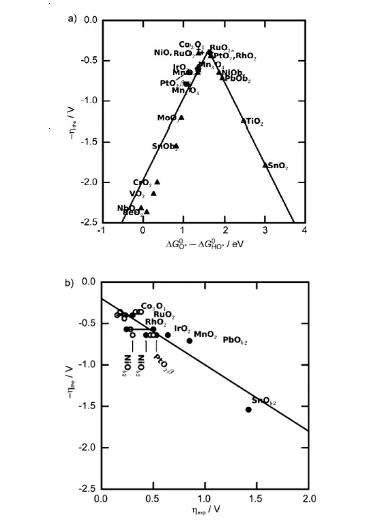
图8的火山图展示了氧化物催化剂在析氧反应(OER)中的理论过电位(ηOER)与描述符(ΔGO* – ΔGHO*)之间的关系,数据点覆盖了多种氧化物材料,包括钙钛矿(如SrCoO₃、LaNiO₃)、金红石(如RuO₂、IrO₂)以及尖晶石(如Co₃O₄)等。
图中活性趋势呈典型的火山形分布,顶点附近的理论过电位最低(约0.4-0.5 V),对应最优的ΔGO* – ΔGHO*值(约1.6 eV)。
具体来看,SrCoO₃位于火山顶点附近,其描述符值为1.48 eV,理论过电位较低,与实验测得的碱性条件下高活性一致;RuO₂和Co₃O₄也接近顶点,理论过电位分别为0.52 V和0.48 V,但实验数据显示Co₃O₄的过电位略高于RuO₂,可能与材料非化学计量比或粒径效应有关。
火山左侧(ΔGO* – ΔGHO* 吸附过强,过电位由HOO形成步骤(ΔG₃)主导;右侧(ΔGO* – ΔGHO* >1.6 eV)的材料(如LaCuO₃)则因O吸附过弱,过电位由HO氧化步骤(ΔG₂)决定。
此外,标度关系(ΔGHOO* ≈ ΔGHO* + 3.2 eV)导致所有数据点沿火山曲线分布,表明HOO与HO的吸附能差固定为3.2 eV,限制了最低理论过电位(约0.4 V)。
图中还显示,某些材料(如NiO、SnO₂)的理论与实验过电位存在偏差,可能与表面非化学计量比或实际反应路径的复杂性有关。总体而言,图8通过数据点的分布验证了描述符的普适性,并量化了不同氧化物催化剂的活性差异,为理性设计OER催化剂提供了明确的热力学依据。
案例2
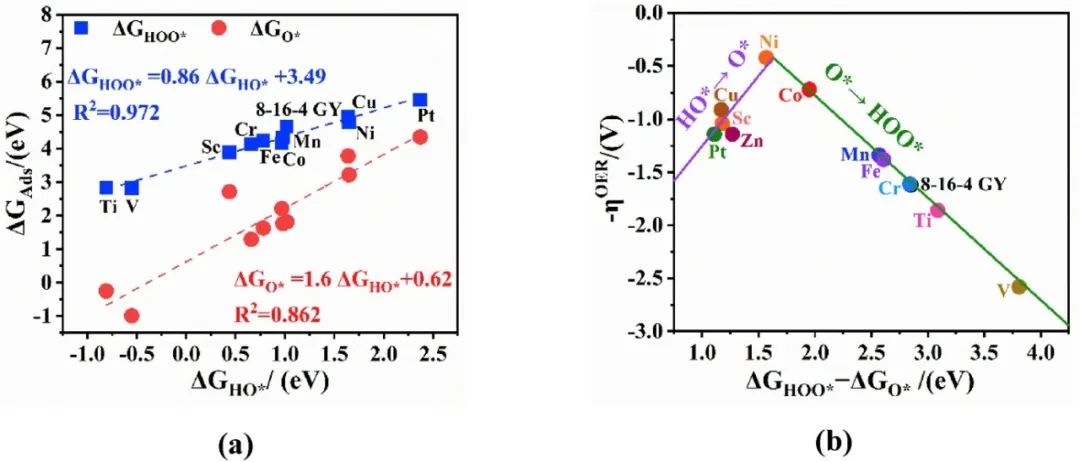
下图中的火山图展示了基于8-16-4 graphyne(GY)负载的单原子催化剂(TM-GY)在析氧反应(OER)中的理论过电位(ηOER)与描述符(ΔGHOO* – ΔGO*)之间的关系。
数据点涵盖了从Sc到Zn的3d过渡金属(TM)以及Pt单原子锚定的GY体系。火山图的顶点对应最低的理论过电位(约0.40 V),描述符值约为1.63 eV,表明此处的催化剂具有最优的OER活性。
具体来看,Ni-GY和Co-GY分别位于火山顶点的两侧,其描述符值分别为1.57 eV和1.95 eV,对应的过电位分别为0.42 V和0.72 V,显示出较高的催化活性。
相比之下,Ti-GY和V-GY的描述符值较低(分别为3.69 eV和3.81 eV),过电位较高(1.86 V和2.58 V),表明其OER活性较差。
火山图的左侧(ΔGHOO* – ΔGO* >1.63 eV)代表HOO形成步骤(ΔG3)为决速步,而右侧(ΔGHOO* – ΔGO* 2)为决速步。
此外,火山图中还显示了ΔGHOO*与ΔGHO*之间的标度关系(ΔGHOO* = 0.86ΔGHO* + 3.49,R²=0.972)以及ΔGO*与ΔGHO*之间的关系(ΔGO* = 1.6ΔGHO* + 0.62,R²=0.862),这些关系进一步验证了描述符的普适性。
值得注意的是,Ni-GY的描述符值最接近理论最优值(1.63 eV),其过电位仅为0.42 V,显著低于其他TM-GY体系(如Sc-GY的1.04 V、Cr-GY的1.61 V、Pt-GY的1.14 V等),表明其在OER中具有优异的催化性能。
此外,Co-GY虽然偏离顶点较远,但其过电位(0.72 V)仍低于许多传统催化剂(如Pt(111)的1.13 V)。
总体而言,通过火山曲线清晰地量化了不同TM-GY催化剂的OER活性差异,并揭示了描述符与过电位之间的非线性关系,为筛选高效OER催化剂提供了理论依据。
案例3
图a展示了TM-PDY催化剂中过渡金属(TM)的d带中心(εd)与OH吸附自由能(ΔGOH)之间的关系,数据点涵盖了Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Ru、Rh、Pd、Os、Ir和Pt等15种金属单原子锚定的PDY体系。
εd值随d电子数的增加呈现周期性变化,3d金属的εd趋势为Sc (0.09 eV) > Ti (0.08 eV) > Cr (-0.51 eV) > Mn (-1.01 eV) > Fe (-1.03 eV) > Co (-1.29 eV) > Ni (-2.53 eV) > Cu (-3.21 eV),而V (0.30 eV)例外。
εd与ΔGOH呈负相关,表明εd越负,TM与OH的结合越弱。例如,Sc-PDY和Ti-PDY的εd接近费米能级(正值),其ΔGOH*分别为-1.23 eV和-0.81 eV,表现出过强的OH*吸附;而Cu-PDY的εd为-3.21 eV,ΔGOH*为1.64 eV,吸附过弱。
Ir-PDY和Rh-PDY的εd分别为-1.57 eV和-1.68 eV,ΔGOH*接近理想值1.23 eV(Ir: 1.19 eV,Rh: 0.86 eV),因此表现出优异的OER/ORR活性。
图b展示了TM-PDY中TM-OH相互作用的积分晶体轨道哈密顿布居(ICOHP)与ΔGOH*的线性关系(R²=0.94),ICOHP越负,TM-OH结合越强。
例如,Sc-PDY和Ti-PDY的ICOHP分别为-5.12 eV和-4.89 eV,对应ΔGOH*为-1.23 eV和-0.81 eV,表明强吸附;而Cu-PDY的ICOHP为-2.01 eV,ΔGOH*为1.64 eV,吸附较弱。
Ir-PDY和Rh-PDY的ICOHP分别为-3.45 eV和-3.67 eV,ΔGOH*适中(Ir: 1.19 eV,Rh: 0.86 eV),验证了其适中的结合强度。
此外,同一周期内金属的ICOHP随d电子数增加而逐渐减小(如3d金属:Sc > Ti > Cr > Mn > Fe > Co > Ni > Cu),进一步证实了电子结构对吸附强度的调控作用。
结合图5和图5可知,εd和ICOHP均能有效描述TM-PDY的催化活性起源:εd反映了金属d电子与O-2p轨道的杂化程度,而ICOHP量化了TM-OH键的强度。Ir-PDY和Rh-PDY因适中的εd和ICOHP值,其ΔGOH*接近理想值,从而表现出低OER过电位(Ir: 0.45 V,Rh: 0.34 V)和ORR过电位(Ir: 0.37 V,Rh: 0.47 V)。
其他体系如Sc/Ti-PDY(强吸附)和Cu/Pd-PDY(弱吸附)则因偏离最优ΔGOH*而活性较差。这些数据为设计高效双功能催化剂提供了明确的电子结构描述符。