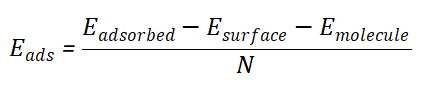VASP(Vienna Ab initio Simulation Package)是一种广泛应用于材料科学和化学领域的第一性原理计算软件,尤其在计算吸附能、电子结构、动力学行为等方面具有重要应用。
VASP通过密度泛函理论(DFT)方法,结合平面波基组和赝势方法,能够高效地计算材料的电子结构和物理性质。华算科技朱老师发现在吸附能计算中VASP提供了多种方法和技巧,以确保计算的准确性和效率。
吸附能是衡量吸附物种与表面之间相互作用强度的重要参数,通常通过计算吸附前后的能量差来确定。在VASP中,吸附能的计算通常包括以下几个步骤:
首先需要对吸附体系进行结构优化,以找到吸附构型的最低能量状态。这通常涉及对吸附位点、吸附物种的几何结构进行弛豫,以确保体系达到稳定的电子结构。
在结构优化完成后,计算吸附体系的总能量。吸附能通常定义为吸附后体系的能量减去吸附前体系的能量,再除以吸附物种的原子数。即:
其中,Eadsorbed 是吸附后的总能量,Esurface 是表面的总能量,Emolecule 是吸附物种的总能量,N 是吸附物种的原子数。
在VASP计算中,需要合理设置输入文件(如INCAR、POSCAR、POTCAR、KPOINTS)以确保计算的准确性和效率。
例如,KPOINTS文件用于指定k点网格,通常采用Monkhorst-Pack网格以提高计算精度;INCAR中设置ISIF=2或3以允许结构弛豫,EDIFF设为1e-4以控制收敛精度。
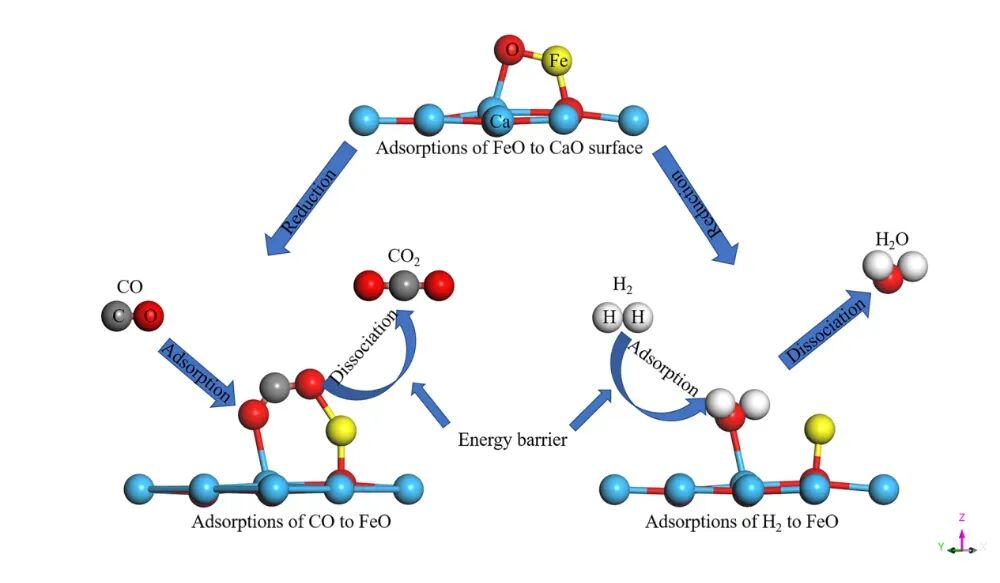
在VASP中,吸附构型的处理涉及多个方面,包括吸附位点的选择、初始构型的获取、结构优化等。
吸附位点的选择对吸附能的计算至关重要。常见的吸附位点包括top、bridge、fcc、hcp等。在实际计算中,可以通过查阅文献或数据库获取已知的吸附位点信息,或通过初步计算确定最优吸附位点。
初始构型的获取可以通过多种方法实现,包括查阅文献、数据库查询或通过简单模型计算。例如,可以通过优化气相分子与表面的结合结构来获得初始构型。
在确定初始构型后,需要对吸附体系进行结构优化,以找到最低能量状态。这通常涉及对吸附位点、吸附物种的几何结构进行弛豫,以确保体系达到稳定的电子结构。
在计算完成后,需要对计算结果进行处理和分析。例如,通过vaspview等工具进行数据提取和分析,以简化计算结果的处理过程。
在VASP计算中,可能会遇到一些常见问题,如能量不收敛、计算效率低等。针对这些问题,可以采取以下解决方案:
如果计算过程中能量不收敛,可能需要调整收敛参数,如减小EDIFF值或增加混合步数(如MAXMIX)。
为了提高计算效率,可以优化k点网格,选择合适的k点密度,以平衡计算精度和计算时间。
在计算过程中,需要确保输入文件的参数设置合理,如ENCUT、K-mesh等参数的设置应与计算体系的性质相匹配。
在吸附能计算中,还有一些高级技巧可以提高计算的准确性和效率:
近年来,机器学习方法在吸附能预测中得到了广泛应用。通过训练机器学习模型,可以快速预测吸附能,提高计算效率。
通过计算差分电荷密度,可以分析吸附物种与表面之间的电子相互作用,进一步理解吸附机制。
结合第一性原理计算和分子动力学模拟,可以更全面地研究吸附行为和动力学过程。
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!