

说明:本文介绍了吸附机理的基本概念、类型及其在催化、能源和环境领域的应用,重点阐述了密度泛函理论(DFT)、分子动力学(MD)和从头算分子动力学(AIMD)等计算方法在解析吸附微观机制中的作用。
读者可系统学习到吸附过程的电子结构、动态行为和影响因素,了解如何通过计算模拟优化催化剂设计和材料性能,为科研人员和工程师提供理论指导,推动能源转化、环境治理等领域的技术创新。
吸附机理的基本概念

吸附是指吸附质从流体相(气体或液体)转移到固体表面的过程,其机理取决于吸附质与表面间的相互作用力。根据作用力性质,吸附分为:
物理吸附:由范德华力或氢键驱动,吸附热低(5-40 kJ/mol),可逆,常形成多层吸附。
化学吸附:涉及化学键(如共价键、配位键)形成,吸附热高(40-800 kJ/mol),单层,通常不可逆。
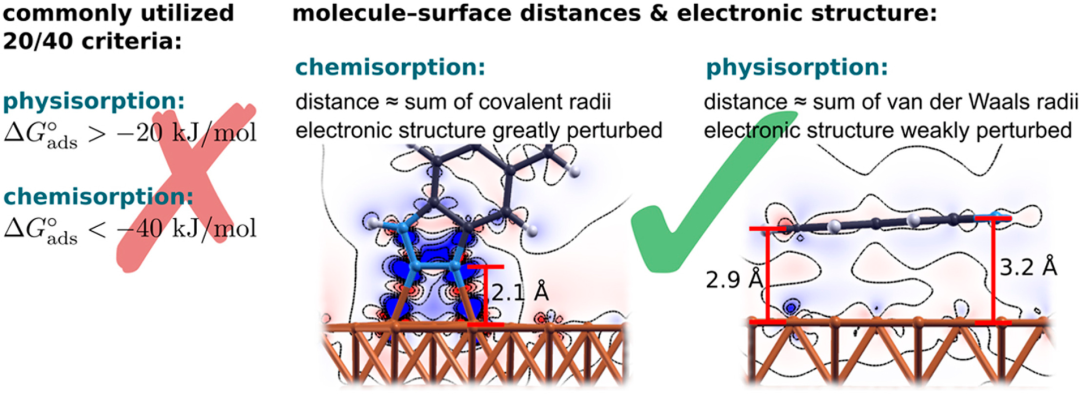
吸附机理的核心是表面活性位点,如缺陷、台阶或低配位原子,这些位点因高表面能而促进吸附。Langmuir吸附等温式(θ = Kp / (1 + Kp),θ为表面覆盖度,K为吸附常数,p为分压)描述单层吸附的平衡行为,揭示吸附饱和现象。
BET模型进一步扩展到多层吸附,公式为 p / [v(1 – p/p0)] = 1/(vm C) + (C-1)p/(vm Cp0),其中v为吸附量,vm为单层容量,C为常数,用于解析物理吸附的层状机理。
影响吸附机理的因素
吸附机理受多种因素调控,可通过计算方法量化分析:
表面电子结构:表面原子的电子密度和能级匹配决定吸附强度。低配位原子(如金属表面的缺陷位点)具有高电子密度,增强化学吸附。
几何构型:表面晶面、孔隙和缺陷影响吸附选择性。多孔材料的孔径限制大分子进入,而特定晶面优先吸附特定分子。
环境条件:温度、压力和pH值改变吸附行为。高温降低物理吸附,压力增加促进气体吸附,pH值影响表面电荷。
计算方法在吸附机理中的应用
计算方法是解析吸附机理的核心工具,能够提供原子尺度的洞见。以下为三种主要方法:
密度泛函理论(DFT)
DOI: 10.1016/j.jhazmat.2019.121997
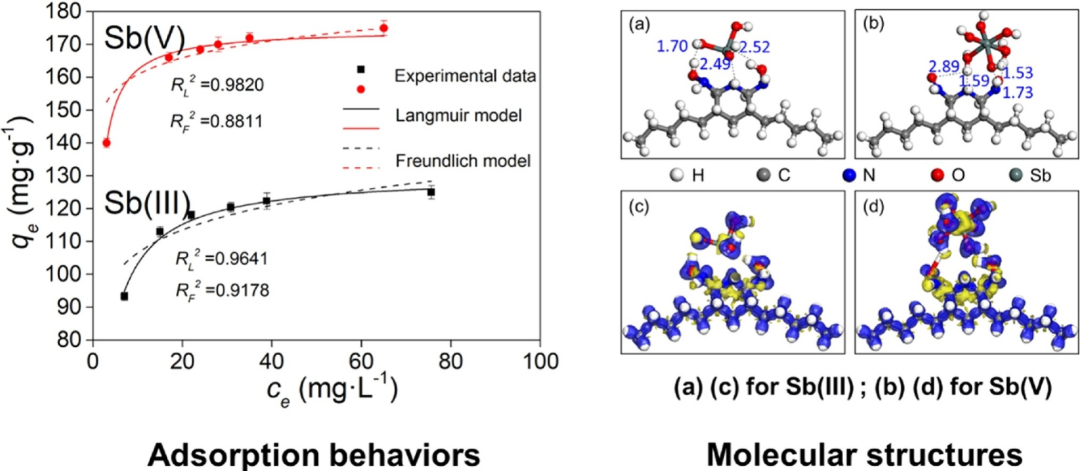
DFT通过计算体系的电子结构,解析吸附质与表面间的电子相互作用机制。它能够描述吸附过程中电荷转移、化学键形成及能量变化,帮助理解物理吸附中弱相互作用的稳定性或化学吸附中轨道杂化的本质。
例如,DFT进一步解释说,UAPAN上Sb(V)的吸附优于Sb(III)的原因是更高的吸附能,更有利的键长和原子电荷密度。因此,UAPAN有望成为从水环境中去除锑的引人注目的候选物,除此之外,DFT还可预测不同位点的选择性,指导催化剂设计。
分子动力学(MD)
DOI: 10.1016/j.jhazmat.2020.123324
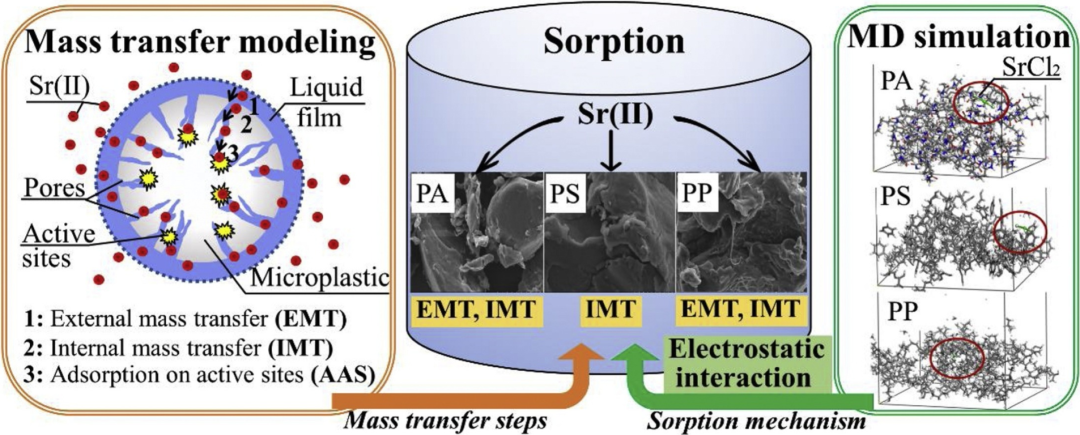
MD基于经典力学,模拟吸附质在表面的动态行为,揭示吸附过程中的分子运动、扩散和构型变化。它能够捕获温度、压力和溶剂对吸附的影响,阐明物理吸附中分子取向的动态平衡或化学吸附中表面原子的重排机制。
例如,微塑料(MP)正在成为全球环境中无处不在的污染物,它们可能会吸附水生环境中的金属离子,造成不良后果,而MD 研究表明,主要吸附机制是静电相互作用。PA-SrCl2、PS- SrCl2 和 PP- SrCl2 的相互作用能分别为−5.638、−6.418 和−13.05 kcal mol−1。
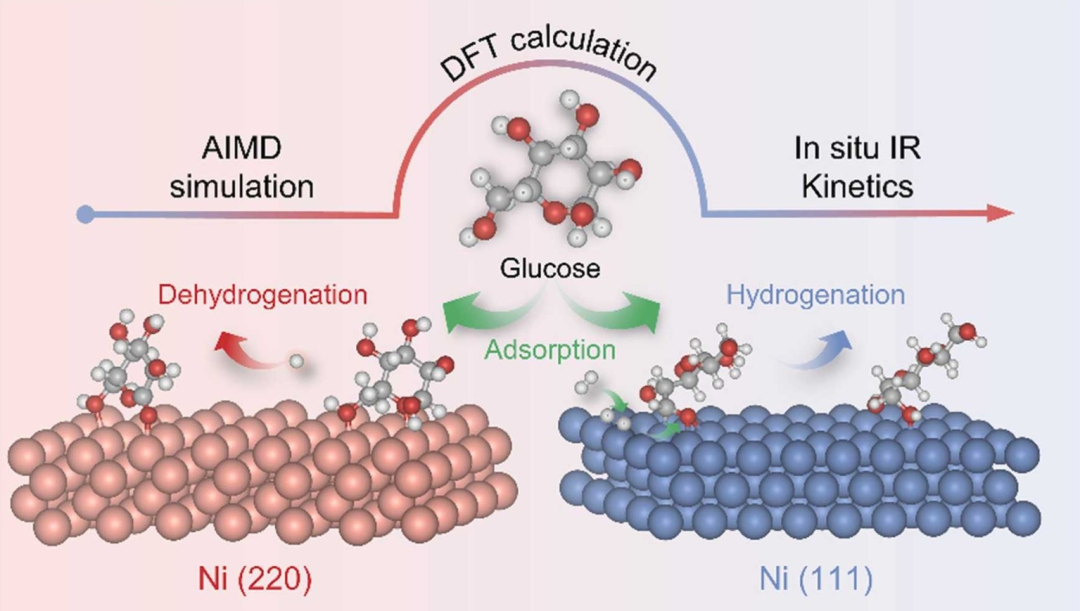
从头算分子动力学(AIMD)
DOI: 10.1016/j.apcata.2023.119462
AIMD结合量子力学和经典动力学,实时模拟吸附过程中的电子和原子动态,特别适合研究化学吸附中的键断裂与生成。它能够揭示吸附质在表面激活的微观路径,如分子解离或表面重构的动态过程。AIMD还可分析反应速率和选择性,为复杂吸附机理提供深入洞见。
结论
吸附机理揭示了吸附质与固体表面的物理或化学相互作用,依赖DFT、MD和AIMD等计算方法进行表征。这些工具通过模拟电子结构、动态行为和能量变化,解析吸附的微观本质。
吸附机理的研究不仅深化了理论理解,还为催化、能源和环境领域的技术创新提供了支持。未来,计算方法的进步将进一步推动吸附机理的精确解析和应用。