

说明:本文华算科技系统介绍了吸附能的基本概念、分类及其在表面科学与催化研究中的核心作用,重点阐述了物理吸附与化学吸附的机制差异、能量特征及其对材料表面反应过程的关键影响。
通过结合密度泛函理论(DFT)的不同泛函选择、范德华力修正及能量校正方法,深入探讨了吸附能计算的理论框架与实操要点。
文中进一步以Nb掺杂Pt催化剂抗CO毒化等顶刊案例为例,展示吸附能计算在揭示电子结构调控、反应路径优化等方面的实际应用。
读者可全面掌握吸附能作为“能量标尺”在催化材料设计、表界面过程模拟及能源转化器件开发中的指导作用,为从事理论计算、催化化学和材料设计的科研人员提供系统的理论支撑与方法参考。
什么是吸附能
吸附能(Adsorption Energy)是表征吸附物与基底结合强度的核心物理量,定义为吸附体系总能量与孤立组分能量之差。
其数学表达式为 Eads = E(slab+adsorbate) – E(slab) – E(adsorbate),其中 E(slab+adsorbate) 代表吸附物与基底形成的复合体系的基态总能量,E(slab) 为未吸附时基底的基态能量。
E(adsorbate) 则是游离态吸附物的基态能量。吸附能值为负值,表示吸附过程是放热反应,数值越负表示吸附作用越强。
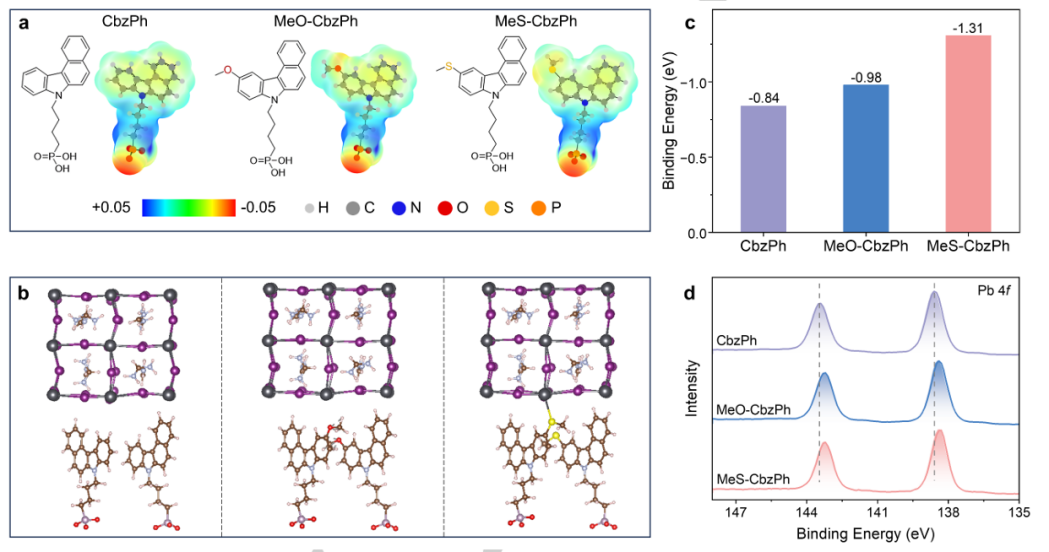

DOI: 10.1002/anie.202419375
吸附过程可分为物理吸附和化学吸附两种类型。物理吸附主要依靠范德华力,吸附能较小(通常几kJ/mol至几十kJ/mol),吸附分子容易脱附;
化学吸附则涉及化学键的形成,吸附能较大(可达上百kJ/mol),被吸附物质即使脱附也可能已发生化学变化。在催化研究中,化学吸附尤为重要,因为大多数催化剂通过化学吸附方式起作用。
吸附能的计算方法
密度泛函理论(DFT)计算
密度泛函理论(DFT)是计算吸附能的主流方法,通过求解Kohn-Sham方程获得体系总能量,适用于处理周期性表面模型。
DFT计算效率较高,能在原子尺度描述吸附物与基底的电子相互作用。然而,DFT计算的核心挑战在于交换关联泛函(XC)的选择,不同泛函对吸附能的计算精度影响显著。
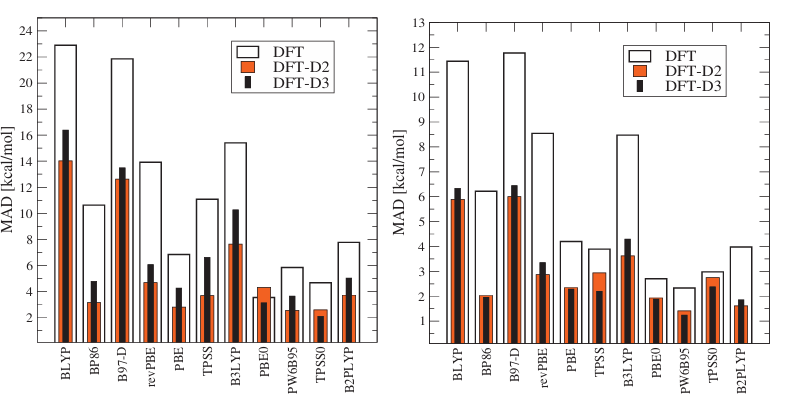
DOI:10.1063/1.3382344
局域泛函(LDA)因过度考虑电子局域性,往往高估吸附能;梯度修正泛函(如GGA-PBE)改善了局域近似的缺陷,但普遍低估吸附能,尤其对涉及范德华力的弱相互作用描述不足。
杂化泛函通过引入非局域交换能提升精度,但计算成本显著增加,仅适用于小体系或高精度验证。
对于弱相互作用(如π-π堆积、氢键、分子物理吸附),需采用专门的范德华力(vdW)修正方法。
DFT-D系列通过添加原子对色散项,基于第一性原理计算色散系数;vdW-DF采用非局域泛函,对长程色散作用的描述更精准;TS-vdW则针对大分子,可避免物理吸附与化学吸附的误判。
计算流程与校正
吸附能计算需要遵循严格的流程和校正步骤。首先需要进行几何优化,确定吸附物在表面的稳定吸附构型。
然后进行能量计算,分别获取复合体系的总能量E(slab+adsorbate)、清洁表面的能量E(slab)及气相吸附物的能量E(adsorbate)。
计算过程中还需要进行关键修正,包括基组叠加误差(BSSE)校正和零点能校正,使能量计算更贴近实际体系。对于高温条件下的吸附,还需要考虑温度效应和熵变对吸附能的影响。
顶刊案例解析
案例背景与研究意义
2025年,青岛大学张连营教授团队研究针对直接甲醇燃料电池阳极催化剂易受CO毒化的问题,通过实验与理论计算相结合的方法,深入探究了Nb掺杂Pt催化剂的抗CO毒化机制。

DOI:10.1021/acs.nanolett.5c02842
直接甲醇燃料电池作为清洁能源技术,其发展受制于阳极催化剂的CO毒化现象。甲醇氧化反应(MOR)过程中产生的CO中间体会强烈吸附在Pt活性位点上,导致催化剂失活。虽然先前研究表明Nb修饰的Pt基催化剂具有较高的抗CO毒化能力,但其内在机制尚不清晰。
研究方法与创新策略
研究团队采用快速焦耳热技术制备了Nb掺杂Pt纳米颗粒,这种方法可实现高温快速合成,确保Nb元素均匀掺杂到Pt晶格中。通过原位ATR-SEIRAS光谱与DFT理论计算相结合,系统研究了Nb掺杂对CO吸附能和反应路径的影响。
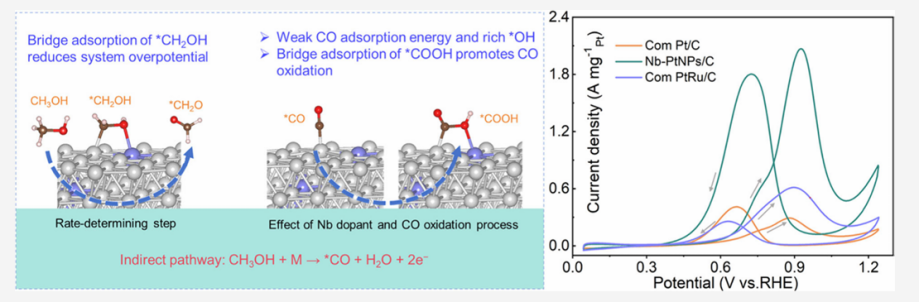
DOI:10.1021/acs.nanolett.5c02842
理论计算部分,研究人员通过密度泛函理论计算了CO在不同表面的吸附能,分析了Nb掺杂对Pt电子结构的影响,特别是d带中心位置的变化。同时,计算了甲醇氧化反应中间物种在催化剂表面的吸附方式和自由能变化,揭示了Nb掺杂促进反应动力学的内在机制。
重要结果与发现
研究结果显示,Nb掺杂显著削弱了CO在Pt表面的吸附能并在较低电位下促进OH物种表面富集。OH物种能够及时氧化去除CO中间体,防止催化剂毒化。同时,Nb的掺入优化了Pt电子结构,使得d带中心下移,减弱了与CO分子的相互作用。
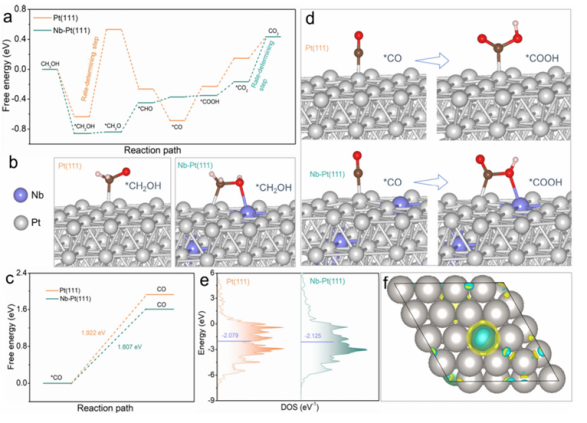
DOI:10.1021/acs.nanolett.5c02842
更重要的是,Nb掺杂形成了CH3OH和COOH物种在催化剂表面的桥式吸附,降低了反应能垒。
电化学测试结果显示,该催化剂甲醇电氧化质量比活性高达2067.2 mA mg-1Pt,是商业化Pt/C催化剂的7.2倍,CO氧化起始电位负移157 mV,同时具有优良的稳定性能。
总结
吸附能作为表征吸附物与基底结合强度的核心物理量,在表面科学、催化化学和材料设计中具有至关重要的作用。从催化反应设计到材料稳定性评估,从高通量筛选到工业应用,吸附能作为“能量标尺“连接着微观结构与宏观性能,推动了材料研究从经验探索向理性设计的转变。
随着计算方法的不断进步和实验技术的创新,吸附能研究将继续深化我们对表面过程的理解,助力新材料设计和性能优化。从原子尺度理解到宏观性能调控的全链条创新,吸附能作为表面科学的“语言“和“密码“,将继续在科学研究和工业应用中发挥不可替代的作用。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服
务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???