

什么是吸附能和结合能
吸附能:指分子在表面或界面上的吸附过程中,单位物质所释放的能量。它反映了吸附分子与表面或吸附剂之间的相互作用力。吸附能越大,表示吸附作用越强,分子越难以脱离表面。
结合能:通常指物质或分子内部的稳定性,尤其是在化学反应或物理过程中形成的化学键所释放的能量。结合能反映了分子或原子之间的稳定性,结合能越大,分子越稳定。
吸附能与结合能的区别
1、作用对象的差别
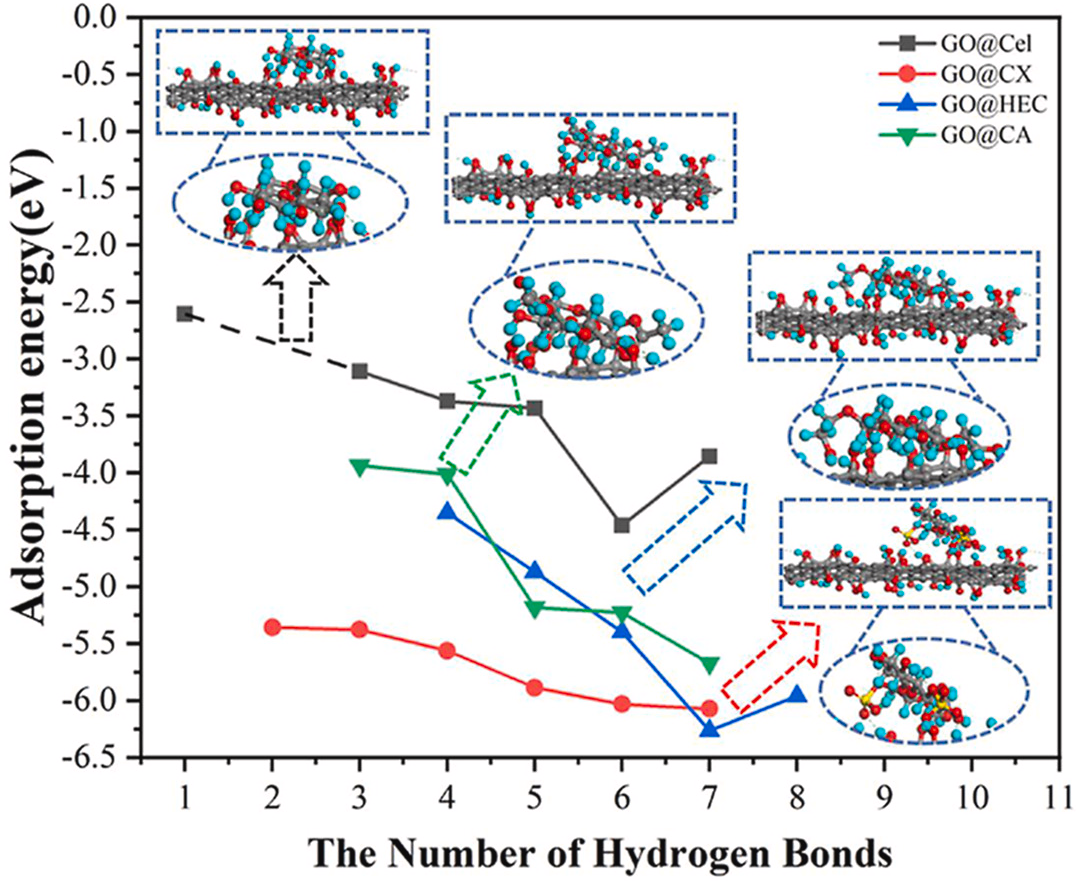

DOI: 10.1016/j.compscitech.2021.109209
吸附能主要描述分子与表面或界面之间的相互作用,特别是分子在表面上的稳定性。在催化反应中,吸附能决定了反应物是否能够有效吸附到催化剂表面并参与反应。例如,以纳米纤维素为基体,以氧化石墨烯纳米片为功能相和增强相,可以生成高性能的生物质基材料。
第一性原理计算已用于确定纤维素衍生物(纤维二糖、黄原酸纤维素、羟乙基纤维素和醋酸纤维素)与氧化石墨烯(GO) 之间的界面结构和相互作用机制。
结果表明,复合体系主要通过氢键和范德华相互作用建立;纤维素/衍生物和GO的种类通过形成各种空间效应和氢键网络对复合体系的稳定性起着重要作用;纤维素衍生物倾向于与 GO 形成更多的氢键,产生大量的电荷转移和更大的吸附能。
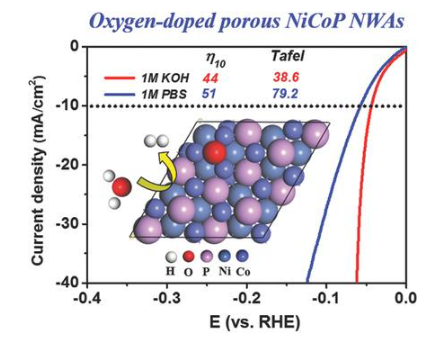
DOI: 10.1002/smll.201800421
结合能则描述分子内部的化学键稳定性,反映分子内原子之间的结合力。结合能越大,分子越稳定。以水分子为例,最佳的氧掺杂NiCoP(≈0.98%氧气)纳米线阵列被发现表现出显着低的析氢反应(HER)超电势44毫伏,以驱动10毫安厘米和38.6毫伏分解的小塔菲尔斜率和在碱性介质中30小时的长期稳定性,说明NiCoP中的氧掺杂确实可以优化原子氢键能。
2、计算方法的差别
DOI: 10.1002/anie.201907966
吸附能的计算通常依赖于分子吸附到表面后的能量变化。研究人员会计算分子与表面接触前后的能量差,通过量子化学计算来评估吸附能的大小。吸附能的计算需要考虑表面和分子间的电子相互作用以及分子几何变化。例如,在催化剂设计中,计算吸附能可以帮助优化催化剂表面的吸附性能。
DOI: 10.1126/sciadv.1603224
结合能的计算则侧重于分子内部的能量变化。通过计算化学键形成和解离时的能量差,结合能能够反映分子内的稳定性。例如,在水分子的计算中,首先计算水分子形成前的能量,再计算形成后的能量,两者之差即为氢氧键的结合能。结合能的计算通常依赖于量子化学方法,如密度泛函理论(DFT),用于精确计算化学键的形成和断裂过程。
吸附能与结合能的联系
吸附能和结合能虽然各自描述不同的相互作用,但它们在某些情况下是密切相关的。在催化反应中,较强的吸附能可能意味着分子与催化剂表面的结合较强,从而导致反应物在表面稳定地存在,促进反应。然而,过强的吸附能也可能导致分子不容易脱附,影响反应效率。因此,吸附能和结合能的平衡对于催化反应的效率至关重要。
总结
吸附能和结合能是描述分子间相互作用的两个关键量。吸附能主要描述分子与表面之间的相互作用,而结合能则侧重于分子内部的稳定性。尽管两者在作用对象和计算方法上有所不同,但它们在实际应用中常常是相互关联的,尤其是在催化剂设计、气体吸附、药物设计和材料科学等领域。通过理解吸附能和结合能的定义、计算方法及其在不同领域的应用,研究人员能够更好地优化材料设计、提高催化效率,并为分子动力学研究提供重要理论支持。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???