

说明:ReaxFF(反应力场)作为一种创新的分子模拟方法,通过引入动态键序概念,实现了对化学键断裂与形成的原子尺度模拟,显著拓展了分子动力学在反应过程研究中的应用范围。
本文华算科技系统介绍ReaxFF的理论原理、参数化流程、典型应用场景及当前面临的挑战,并展望其与机器学习结合等未来发展方向,为计算化学与材料科学研究提供关键方法支撑。
ReaxFF的核心原理与理论框架
ReaxFF反应力场是一种半经验的分子力场,其设计的初衷是为了以可接受的计算成本模拟大规模复杂体系中的化学反应动力学。
与传统力场最大的区别在于,传统力场通常基于固定的化学键连接关系,将分子视为一组通过弹簧连接的原子,因此无法描述键的生成和断裂。ReaxFF则突破了这一限制,其核心创新在于引入了“键序”(Bond Order)的概念。
键序是一个连续变化的函数,它直接与原子间的距离相关联。当两个原子相互靠近时,它们的键序从0逐渐增加到整数(如1、2、3),代表了单键、双键、三键的形成;
反之,当原子远离时,键序又会平滑地降至0,模拟了化学键的断裂过程。这种动态的成键描述使得ReaxFF能够自然地处理化学反应、相变以及材料损伤等复杂过程。


图1反应分子动力学模拟理论框架
DOI:10.3969/j.issn.2096-8299.2024.03.013
ReaxFF的能量函数是一个复杂的、由多个部分构成的表达式,旨在精确描述原子在形成和断裂化学键过程中的能量变化。系统的总能量E是多个能量项的总和,这些能量项都与键序紧密相关。
主要包括:键能(Ebond),它描述了成键原子的能量;原子过配位能(Eover)和欠配位能(Eunder),用于惩罚偏离理想价态的原子;孤对电子能(Elp);
键角能(Eval)和二面角能(Etor),这些项的能量贡献会随着中心键的键序减弱而消失;以及非键相互作用,包括范德华力(EvdW)和库仑力(ECoulomb)。
范德华力和库仑力在所有原子对之间计算,并通过屏蔽函数在短距离内进行平滑处理,以避免出现能量奇点。正是这种全面的能量描述,使得ReaxFF能够在模拟化学反应时,提供接近量子力学(QM)计算的能量路径和反应势垒。
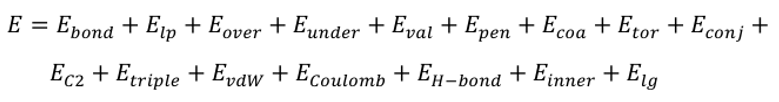
ReaxFF力场的开发与参数化
ReaxFF的强大功能依赖于一套经过精心优化的参数。力场参数化是一个复杂且耗时的工作,其目标是使ReaxFF计算得到的能量、力、电荷等物理量尽可能地与高精度的量子化学计算(通常是密度泛函理论,DFT)数据或实验数据相吻合。
这个过程大致可分为四个关键步骤:数据生成、训练集构建、优化算法选择和验证。首先,研究人员需要构建一个全面的训练数据集,其中包含与目标研究体系相关的各种分子构型和反应路径,例如键的拉伸、键角弯曲、分子反应中间体和过渡态等。
这些数据点的能量、原子受力等信息通过高精度的DFT计算获得。一个高质量的训练集对于力场参数的准确性和普适性至关重要。
在构建好训练集之后,下一步是选择合适的优化算法来拟合ReaxFF中的数十个甚至上百个参数。由于参数空间维度高且高度非线性,传统的梯度下降法容易陷入局部最优。
因此,全局优化算法如蒙特卡洛模拟退火、遗传算法(GA)以及协方差矩阵自适应演化策略(CMA-ES)
被广泛应用。近年来,随着人工智能技术的发展,基于机器学习的参数化框架,如JAX-ReaxFF和INDEEDopt,通过构建代理模型来预测参数与误差之间的关系,极大地提高了优化效率和自动化程度。
整个参数化过程通常在专门的软件工具中完成,例如ParAMS、ASE和pymatgen等用于数据处理和流程控制,而LAMMPS则作为执行ReaxFF计算的核心引擎。
最后,优化得到的力场参数必须通过一个独立的验证集进行检验,以评估其在训练集之外的预测能力和物理真实性。
ReaxFF的应用领域
凭借其模拟化学反应的独特能力,ReaxFF已成为连接微观理论与宏观实验的桥梁,广泛应用于化学、材料科学、物理和生物学的交叉领域。
在能源与燃烧领域,ReaxFF能够模拟碳氢燃料在高温高压下的热解和氧化过程,揭示复杂的燃烧反应网络和关键中间产物的生成路径,这对于设计更高效、更清洁的发动机至关重要。
例如,通过模拟氢气的燃烧,研究人员可以观察到过氧基自由基(HO2)在链式反应中的关键作用,并量化其对反应动力学的影响。这种原子级别的洞察是传统宏观动力学模型难以提供的。
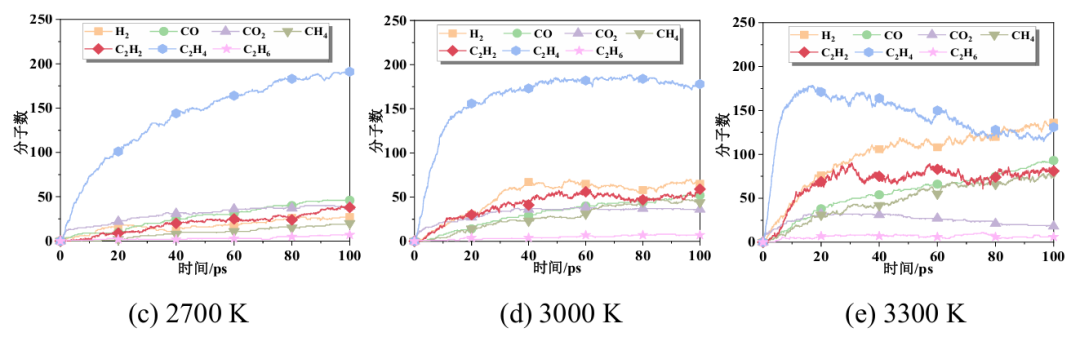
图2 三元混合绝缘油-纤维素复合体系热裂解特征气体最高含量及100 ps内变化情况
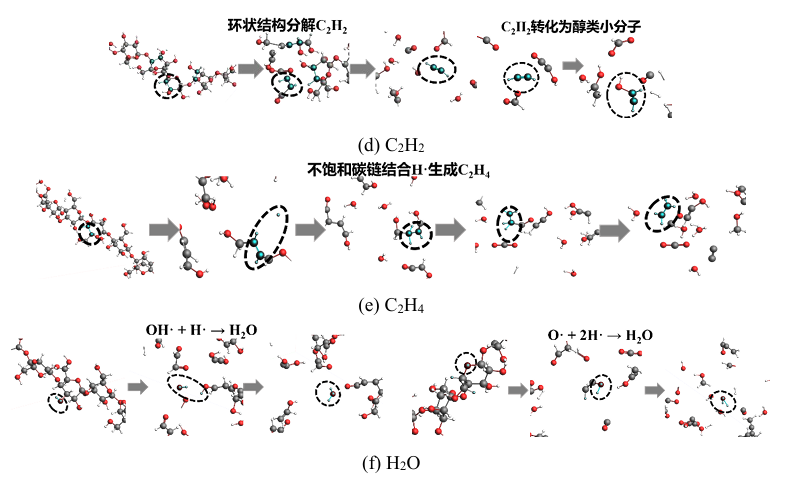
图3 特征气体及H2O主要生成及演化路径
在催化化学中,ReaxFF被用于研究催化剂表面的反应机理。研究人员可以模拟反应物分子在催化剂活性位点的吸附、解离、重组和产物脱附的全过程,从而理解催化剂的活性和选择性来源。
而在电池材料科学中,ReaxFF的应用尤为突出。例如,在锂离子电池研究中,它可以模拟电解液在电极表面的还原或氧化分解过程,揭示固体电解质界面(SEI)膜的形成机理、化学成分和微观结构。
一项具体研究利用ReaxFF详细模拟了碳酸乙烯酯(EC)分子的还原分解路径,为理解和改善电池的循环寿命和安全性提供了原子尺度的理论指导。
此外,ReaxFF还被用于研究聚合物材料的老化与降解、金属材料的应力腐蚀与断裂、以及生物大分子的相互作用等众多前沿课题。
关键挑战与未来展望
尽管ReaxFF取得了巨大成功,但它仍然面临一些关键挑战。首先,力场参数化仍然是最大的瓶颈。开发一套高质量、高普适性的力场参数需要大量的专家知识和计算资源,其过程繁琐且难以自动化。
参数的准确性直接决定了模拟结果的可靠性,而一个力场通常只对其训练集中包含的化学环境有效,对于未知体系的“外推”能力有限。
其次,虽然ReaxFF相比量子化学计算效率极高,但相较于非反应力场,其计算成本仍然要高出一个数量级以上,这在一定程度上限制了其能够模拟的时间和空间尺度。
最后,对于某些复杂的物理化学过程,如长程静电相互作用、电荷转移的精确描述以及量子隧穿效应,目前的ReaxFF框架仍有待改进。
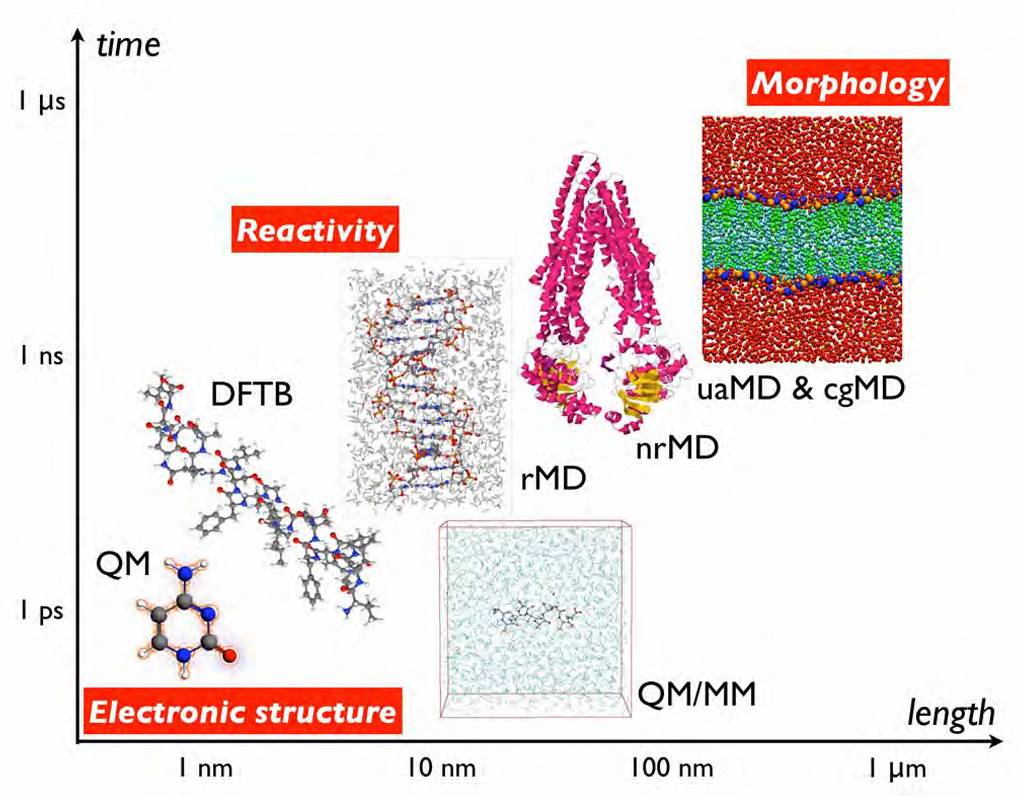
图4 不同时间尺度和空间尺度的分子模拟方法
DOI: 10.1088/0022-3727/49/5/054002
展望未来,ReaxFF的发展将主要集中在两个方向。一是与机器学习的深度融合。利用机器学习模型加速参数优化过程,甚至直接构建基于神经网络的反应势函数,有望从根本上解决参数化的瓶颈问题。
通过主动学习等策略,机器可以自动探索化学空间,智能地生成新的训练数据,从而开发出更准确、更具普适性的力场。
二是扩展其物理模型的广度与深度。例如,开发能够描述电子极化效应的eReaxFF,或者将力场与蒙特卡洛方法结合以模拟更长时间尺度的事件。
随着计算能力的不断增强和算法的持续创新,ReaxFF及其后续发展方法将在揭示从生命起源到新材料设计的各类复杂化学反应机理中,扮演越来越重要的角色,继续充当连接微观量子世界与宏观现实世界的关键桥梁。
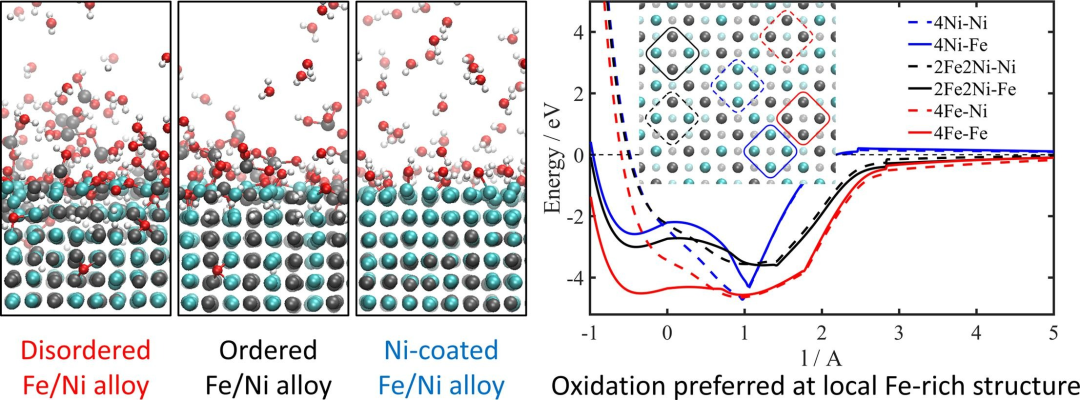
图5超临界水氧化铁/镍合金表面:ReaxFF分子动力学模拟DOI: 10.1016/j.apsusc.2021.149519
总结
ReaxFF反应力场凭借其动态键序机制,有效弥合了量子计算与经典分子动力学之间的尺度鸿沟,成为研究化学反应、材料降解、能源转化等过程的重要工具。
其在燃烧机理、电池界面和催化反应等领域的应用已展现出强大解释与预测能力。尽管在参数化和计算效率方面仍存在挑战,随着机器学习融合和多尺度方法发展,ReaxFF有望在复杂体系模拟中发挥更大作用。