




什么是分子动力学(MD)模拟?
分子动力学(MD)模拟是一种基于经典力学的计算方法,通过求解牛顿运动方程,模拟分子、原子或离子在特定时间和空间中的运动轨迹。它以皮秒(10⁻¹²秒)到微秒(10⁻⁶秒)的时间分辨率和埃(10⁻¹⁰米)到纳米的空间分辨率,捕捉物质的微观动态,如分子间相互作用、扩散路径和结构演化。
MD模拟的核心是将分子体系视为由粒子组成的集合,通过力场(描述粒子间相互作用的数学模型)计算每个粒子的受力、速度和位置,生成动态轨迹。

图1.大分子晶体的分子动力学。DOI:10.1002/wcms.1402
MD模拟的历史可以追溯至20世纪50年代,首次由Alder和Wainwright用于研究硬球模型的相变行为。随着计算能力的提升,MD模拟从简单的气体模型扩展到复杂的生物分子、材料和化学体系。
1977年,Karplus等人首次将MD模拟应用于蛋白质研究,开启了其在生物学领域的广泛应用。如今,MD模拟已成为多学科交叉的基石技术,广泛用于化学、物理、生物学和材料科学。
为何MD模拟如此重要?
传统实验技术(如X射线晶体学、核磁共振、红外光谱)在解析分子结构和动态行为时,常常受限于时间分辨率(毫秒级)或空间分辨率(纳米级)。MD模拟通过计算弥补了这些局限,提供以下关键信息:
分子间相互作用:揭示范德华力、氢键、静电相互作用和π–π堆积等微观机制。
动态过程:模拟分子扩散、蛋白质折叠、材料断裂和化学反应等动态行为。
物理化学性质:预测扩散系数、热导率、溶剂化能、结合自由能等关键参数。
例如,在药物设计中,MD模拟可揭示药物分子与靶蛋白的结合路径;在材料科学中,它能优化纳米材料的力学和导电性能;在储能领域,它能指导电解液的离子传输优化。通过MD模拟,我们能在分子级别进行设计和优化,大大加快新技术和新材料的开发进程。
MD模拟的基本原理
MD模拟的核心是基于经典力学,通过力场计算粒子间的相互作用力。常用力场包括AMBER、CHARMM和OPLS-AA,这些力场通过势能函数描述分子间的键合(如键长、键角)和非键合(如范德华力、静电)相互作用。模拟过程包括以下步骤:
体系构建:定义模拟对象(如蛋白质、电解液、纳米材料),设置初始位置和速度。
力场选择:根据体系特性选择合适的力场模型。
运动方程求解:使用数值积分(如Verlet算法)计算粒子运动轨迹。
数据分析:提取关键参数,如径向分布函数(RDF)、均方位移(MSD)和结合自由能。
模拟通常在恒温恒压(NPT)或恒温恒体积(NVT)条件下进行,使用软件如GROMACS、LAMMPS或NAMD等,结合高性能计算(HPC)实现大体系模拟。
MD模拟的核心应用
1、生物分子动力学:解析蛋白质折叠与药物结合
MD模拟在生物分子研究中已成为解析蛋白质折叠、酶–底物相互作用及药物–靶点结合机制的关键工具。近年来的研究进一步强调了水合壳动力学在这些生物过程中的核心作用,为精准医疗和药物开发提供了更深入的理论与机制支持。
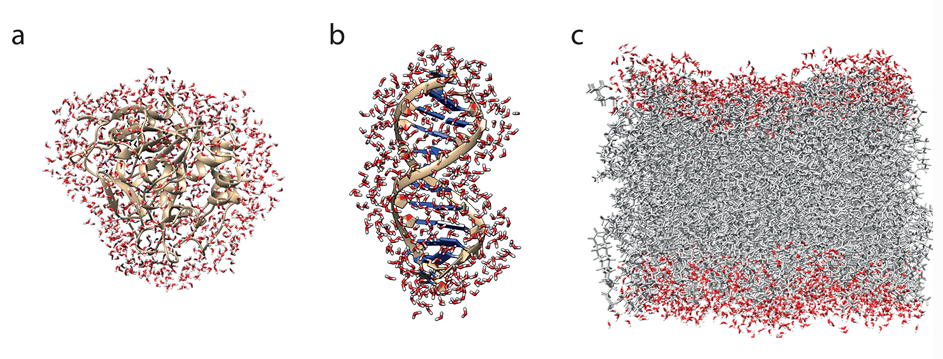
图2.(a)蛋白质、(b)DNA 双链以及(c)磷脂双层的典型水合鞘。DOI:10.1021/acs.chemrev.6b00765
核心机理:
蛋白质折叠和药物结合不仅由分子间的直接弱相互作用(如氢键、疏水作用等)驱动,更显著地受到周围水合水动态行为的调控。水分子通过其氢键网络的快速重排(发生在皮秒时间尺度)和重新取向(约2 ps),起到“润滑”生物大分子构象运动的作用,并主动参与分子识别与结合事件。
MD模拟通过构建包含显性水分子(explicit water)的蛋白质–溶剂体系,不仅能追踪氨基酸残基的运动轨迹,还可直接揭示水合壳的结构涨落、氢键寿命及其空间异质性。
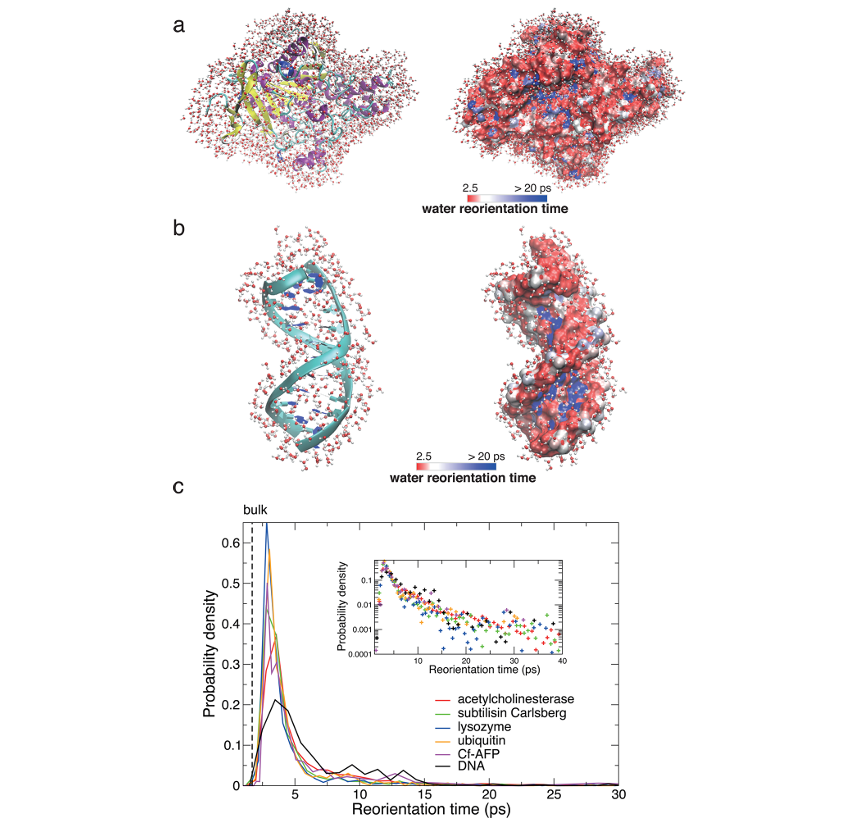
图3.(a)乙酰胆碱酯酶及其水合壳层水分子(左),以及壳层中水分子重定向动力学图(右);(b)围绕 DNA 十二聚体的情况;(c)一系列球状蛋白质 57,61 以及 DNA 58 附近水分子重定向时间的概率密度,采用线性标度和半对数标度(插图)。DOI:10.1021/acs.chemrev.6b00765
药物设计应用:
在药物设计领域,MD模拟常与MM/GBSA或MM/PBSA等方法结合,用于计算结合自由能(ΔG),其中极性溶剂化能——与水合壳密切相关的部分——是评估结合亲和力的关键组成。模拟还能揭示药物分子如何扰动靶点蛋白表面的水合动力学,进而指导药物的优化以提高选择性和效力。
2、材料科学:优化纳米材料与界面特性
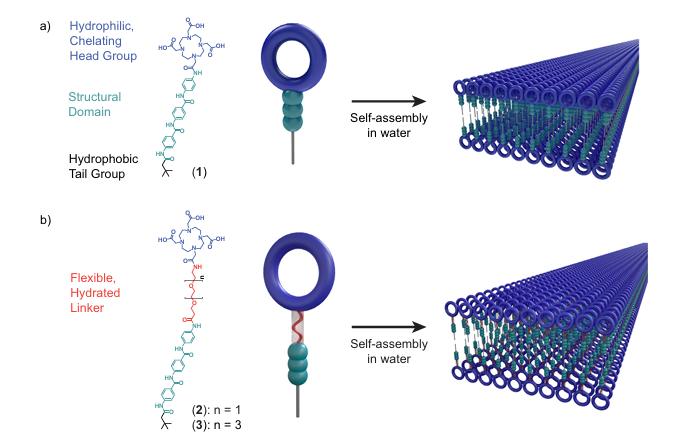
图4.超分子组装体所具有的可调表面化学特性能够控制表面动态和水合。DOI:10.1038/s41467-024-51494-4
MD模拟揭示了界面动力学在合成材料表面化学事件中的关键作用,特别是在水合环境和表面柔性的影响下。研究发现,表面装饰的金属螯合剂在水合和表面动力学增强的情况下,其结合能力显著提高。
通过引入短链的乙烯基醚(OEG)连接子,可以显著增加表面螯合基团的构象流动性,并促进其与周围水分子的相互作用。这一动态行为的增强使得螯合剂在表面上的结合亲和力提高。
本研究中,使用分子动力学(MD)模拟结合电子顺磁共振(EPR)技术来探究不同长度的OEG连接子对表面动力学的影响。通过MD模拟,研究了不同结构的自组装纳米结构的表面水合动力学和柔性对螯合性能的影响。通过此类模拟,科学家可以进一步优化材料的表面化学特性,以实现更高的性能和功能。
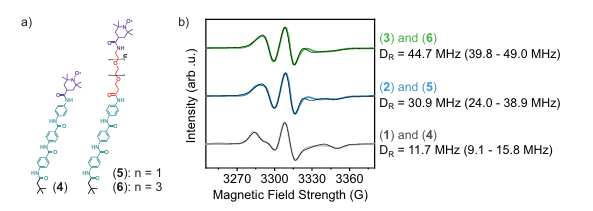
图5. 界面材料和水动力学通过引入柔韧的、水合的表面连接剂得以调节。DOI:10.1038/s41467-024-51494-4
3、储能技术:优化电解液与电池性能
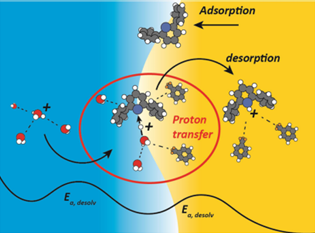
图6.水/薄膜晶体管界面处的 PT 分子图像。DOI:10.1021/jacs.4c18349
MD模拟在储能领域用于研究电解液的离子传输、界面动态及添加剂设计,助力高性能电池的开发。通过模拟电解液中的质子转移过程、离子扩散等,MD模拟能够优化电池性能,推动新型储能技术的研发。
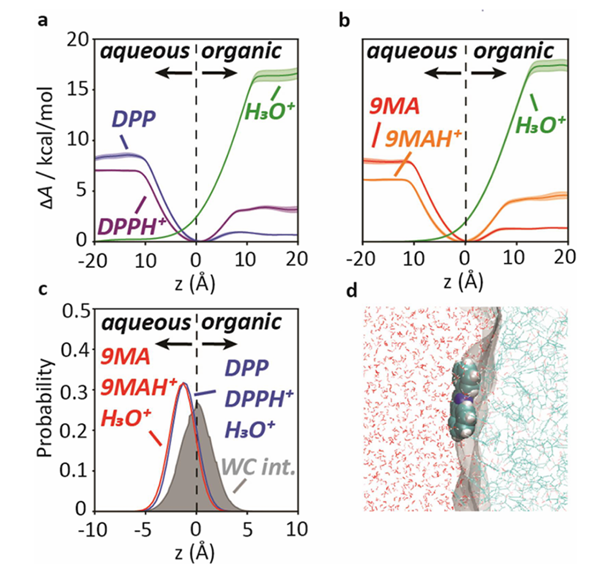
图7.水-TFT 结合界面处 H3O+、DPP、DPPH+、9MA、9MAH+的分子动力学模拟。吉布斯分界面(GDS)位于 z = 0 处,而 z 和 z > 0 分别对应于水相和有机相。DOI:10.1021/jacs.4c18349
质子转移(PT)在电化学界面中是一个基础性过程,广泛应用于水电解、二氧化碳还原和生物催化等领域。通过MD模拟,研究了在不同电解质体系中,质子如何从水相转移到有机相,揭示了质子转移机制的不同路径。
总结
分子动力学(MD)模拟作为一项强大的计算工具,已经成为现代科学研究中的核心技术之一。通过模拟分子、原子或离子的动态行为,MD模拟能够在原子级分辨率上揭示物质的微观运动规律。这项技术不仅在生物分子研究、药物设计、纳米材料优化等领域取得了显著进展,还为储能技术的创新提供了理论支持。
然而,尽管MD模拟展现出巨大的潜力,仍面临计算资源消耗、力场精度等挑战。未来,随着多尺度模拟、人工智能技术的应用和计算能力的提升,MD模拟将在更加广泛的领域中推动科学与技术的突破。