

分子筛是一类重要的多孔材料,因规整的孔道结构、独特的择形(形状选择性)性能和可调控的表面化学性质,在气体催化和吸附分离领域发挥着关键作用。
传统的硅铝酸盐分子筛(沸石)以及新型的金属有机框架(MOF)和共价有机框架(COF)等多种分子筛材料被相继开发,用于CO₂捕集、烃类气体分离以及多相催化等应用。
随着研究的深入,理论模拟手段在揭示分子筛体系的微观机理方面不可或缺,包括密度泛函理论(DFT)、分子动力学(MD)和蒙特卡洛(MC)模拟等,它们能够在分子水平上提供对吸附过程、催化反应路径和孔道内扩散行为的深入认识。
本文将系统综述分子筛的研究背景与结构特性、其在气体吸附和催化中的应用现状,并重点介绍当前主要的理论模拟方法(DFT、MD、MC)在相关研究中的具体应用和优势。
分子筛的结构特性与研究背景
分子筛通常指具有均一微孔结构的结晶材料,可按组成分为无机分子筛(典型为硅铝酸盐沸石)、有机–无机杂化分子筛(如MOF)以及纯有机骨架分子筛(如COF)等类别。
传统沸石分子筛由硅、铝氧四面体通过共顶点连接形成规则的孔道和笼状结构,其孔径一般在纳米尺度,具有高度的结晶度和热稳定性。这些规整孔道赋予分子筛择形效应,使其在催化和吸附中对分子具有尺寸和形状选择性。
例如,沸石分子筛的骨架提供了直径约0.3–0.8nm的微孔道和明确的交叉通道,骨架上的Brønsted酸位(如[Si–O(H)–Al]桥羟基)位于孔内,可作为活性中心决定烃类反应的初始吸附与活化方式。
分子筛孔道的“纳米反应室”效应能通过几何限制使体积庞大的中间体难以形成,从而抑制副反应发生;同时孔壁对特定过渡态提供范德华贴壁稳定作用以降低反应能垒。这种限域效应使分子筛在石油化工中的许多催化过程(如催化裂化、歧化、烷基化等)展现高选择性和活性。
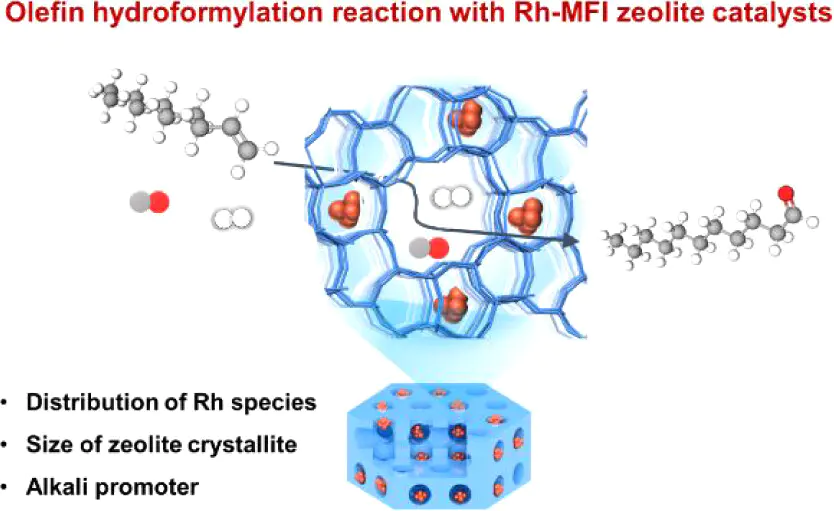

DOI:10.1021/jacs.4c15445.
相比之下,新型的多孔晶体材料MOF和COF具有更丰富的结构多样性和功能可调性。MOF由金属离子/簇与有机配体通过配位键组装而成,呈三维有序网络结构,孔径从微孔到介孔可调,比表面积常超过1000m²/g,某些MOF的比表面积甚至高达10000 m²/g。
MOF因其超高比表面积和高度可调孔径被誉为“分子乐高”。典型MOF如IRMOF-74、ZIF-8、UiO-66等,拥有特定拓扑孔道,能够通过选择合适的金属节点和有机配体,实现孔道表面功能化,例如引入–SO₃H、–NH₂、未配位金属位等,以增强对目标气体的亲和作用或催化活性。
COF则完全由轻元素有机单元通过共价键连接,形成类似石墨层状或三维网状的多孔结构,具备低密度和高孔隙率的特点。总体而言,这些新型框架材料在气体存储(如H₂、CH₄)、CO₂捕集、选择性催化转化等方面展现出巨大潜力。
相较传统沸石,MOF/COF不仅孔径和表面化学可定制,而且某些MOF具有比沸石更高的BET比表面积(例如MIL-101的比表面积约4230m²/g,MOF-210高达6240m²/g)和更大的孔容,这使它们在气体吸附容量上具备显著优势。
需要指出的是,分子筛材料种类繁多,但都共享微孔结构这一核心特征,而孔道结构与表面性能的差异使其在不同气体体系中表现各异。
在气体吸附中的应用
气体吸附与分离是分子筛的重要应用方向之一。凭借微孔提供的巨大内表面积和选择性孔径,分子筛被广泛用于从混合气中选择性地吸附目标气体分子,实现提纯或分离。
例如,在工业上常用的变压吸附(PSA)过程中,低硅铝比的沸石(如13X型、4A型分子筛)由于骨架中存在可交换阳离子位,对极性或易极化气体(如CO₂)具有很高亲和力,因而被优先用于从甲烷、氮气、氢气等混合气中去除CO₂。
研究表明,沸石丰富的阳离子位和不均一表面赋予其强烈的吸附势,使其在低压下即可大量吸附CO₂,从而在合成气、天然气净化等过程中表现出优异的CO₂分离效果。
例如,X型和A型沸石对CO₂的选择性吸附远高于对H₂、N₂等非极性气体,因而在从合成气或天然气中脱除CO₂时常被优选使用。然而,低硅铝沸石在循环再生时可能存在完全再生困难的问题(如PSA减压再生不完全),这在实际应用中需要权衡其吸附容量和再生能耗。
除了传统沸石,金属有机框架等新型多孔材料在气体吸附领域近年来备受关注。MOF材料的孔径和表面功能可通过晶体工程进行精确调控,使其能够针对特定气体分子进行结构优化,从而在某些场合展现优于沸石的吸附性能。
许多MOF具有超高的理论吸附容量,例如MOF-200、MOF-210在25–31℃、高压下对CO₂的饱和吸附量可达30–50mmol/g,显著超过传统沸石和活性炭。
尽管这些高容量通常是在较高压力(数十巴)下获得,在实际PSA工况(约4–6巴)下尚需进一步比较,但MOF的优势在于其可设计性:通过在孔壁引入特定官能团或开放金属位,MOF可以大幅提升对目标气体的选择性。
例如,Mg-MOF-74因孔道中高密度的未配位金属位,对CO₂显示出极高的亲和力和选择性,实验和模拟均证明其在CO₂/N₂、CO₂/CH₄分离中性能优异。
又如,ZIF-8具有类似沸石的结构但孔径更大,对挥发性有机物(VOC如苯、甲苯)的吸附容量高于传统沸石,因为其孔道既能容纳较大分子,又带有有机骨架对有机分子的较强相互作用。
在工业气体纯化、温室气体捕集以及空气净化等领域,分子筛吸附剂(包括沸石、MOF等)已成为不可或缺的核心材料,其吸附等温线、选择性和循环稳定性等性能指标是当前研究的重点。
通过实验测定结合分子模拟预测,人们不断筛选和优化新的分子筛材料,以期满足更高效、更节能的气体分离需求。
在气体催化中的应用
由于具备独特的限域环境和可调酸/碱活性位,分子筛在多相催化领域发挥着举足轻重的作用。酸性沸石分子筛长期以来是石油化工中的关键催化剂:例如,HY沸石用于催化裂化(FCC)将重油转化为汽油馏分,HZSM-5用于甲醇制烯烃(MTO)和烃类芳构化反应,H-beta沸石用于烷烃异构化提高辛烷值等。
在这些反应中,分子筛骨架提供了强Brønsted酸中心,可活化反应物分子(如质子化烯烃或醇),而孔道的空间限制使得反应路径偏向于生成特定尺寸或形状的目标产物(择形催化)。
经典实例包括:HZSM-5的中孔道限制抑制了烯烃的进一步缩合和结焦,从而高选择性地产生轻烯烃和芳香烃;SAPO-34分子筛的小笼型孔道则在MTO反应中促进生成乙烯、丙烯等小分子烯烃并抑制长链副产物的形成。
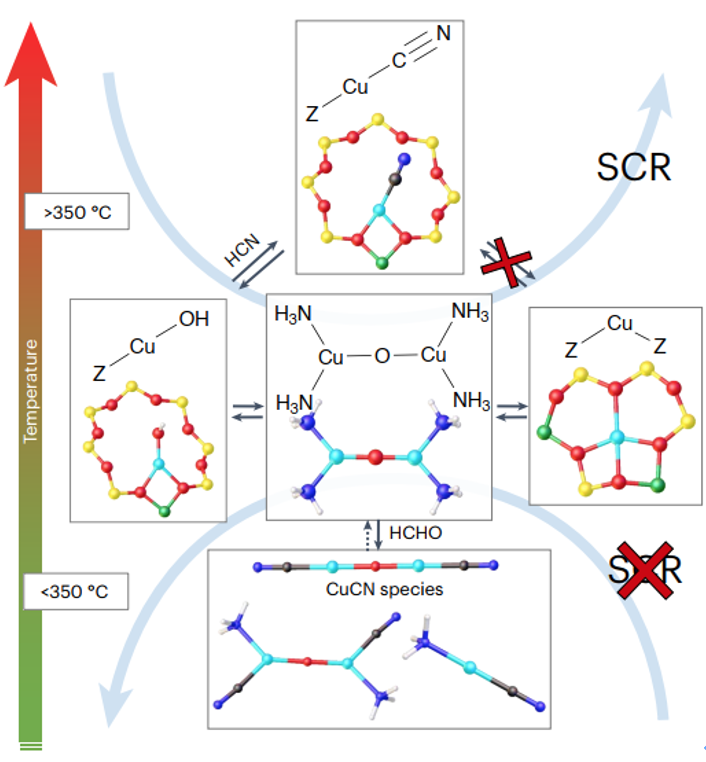
DOI:10.1038/s41929-025-01389-4.
分子筛还在环境催化领域扮演重要角色。例如,Cu或Fe离子交换的分子筛催化剂被用于选择性催化还原(NOxSCR)反应中,以去除燃烧排放中的氮氧化物。
具有CHA拓扑的Cu-SSZ-13(硅铝磷酸盐SAPO-34的硅铝沸石对应物)是商用柴油车尾气NH₃-SCR催化剂,由于其小孔笼结构对NH₃和NOx的优异传质和择形效应,以及Cu²⁺离子作为活性位的高分散性,使其在低温下即可高效地将NOx还原为氮气。
又如,含钛硅分子筛(TS-1)因在MFI骨架中引入了高分散的Ti(IV)活性位,能够在过氧化氢存在下选择性催化丙烯环氧化生成环氧丙烷,是绿色氧化反应催化剂的成功范例。此外,分子筛的孔道还能锚定金属纳米粒子或单原子,从而制备分子筛负载型金属催化剂。
这种催化剂结合了分子筛的择形性和金属的独特活性,例如在分子筛限域空间内引入单原子Pt、Rh等,可实现接近100%原子利用率并避免团聚失活,同时孔壁提供协同作用调控其电子态,提高催化选择性。
近年来,新型MOF和COF材料也逐步用于气相催化反应。一方面,某些MOF自身含有催化活性位(如含未配位金属的节点可以催化Lewis酸反应,或含功能基团的配体参与红ox反应);另一方面,MOF/COF可作为前驱体,在热解后形成具有纳米孔道的金属/碳复合催化剂。
总体来说,在从石油炼制、化工合成到尾气治理的众多气相催化过程中,各类分子筛催化剂凭借孔道限域效应与酸碱可调的优势,实现了高效率和高选择性的催化性能。
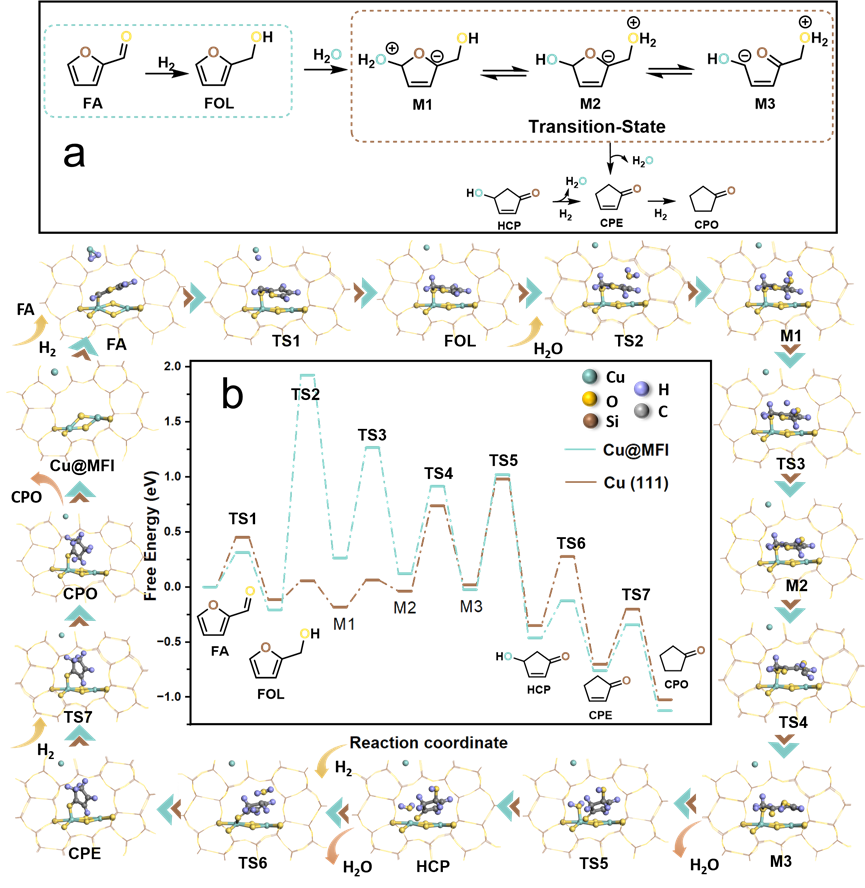
DOI:10.1016/S1872-2067(25)64741-3
理论模拟方法的应用
尽管分子筛在气体吸附和催化中已有广泛应用,但深入理解其微观作用机理并指导材料与工艺优化,离不开理论计算和分子模拟手段的助力。目前,DFT、MD和MC三类模拟方法被广泛用于分子筛相关研究中,各具优势且相互补充。
密度泛函理论(DFT)计算
DFT属于量子化学第一性原理计算方法,可在原子电子水平上解析分子筛内吸附和反应的热力学与动力学性质。在分子筛催化研究中,DFT通常采用周期模型模拟分子筛晶格,从而考察反应物或中间体在骨架酸位上的吸附构型与能量,以及过渡态的结构和反应能垒。
通过对比不同孔道环境下同一活性位的计算结果,研究者能够量化限域微环境对反应的影响,例如比较大孔与小孔分子筛中某反应过渡态的自由能垒差异,从而判断限域效应是否有利于目标反应。
DFT计算还能解析分子筛酸性强度和活性位特征,例如通过计算质子亲和能来评价沸石Brønsted酸酸强,或通过电子态分析(如态密度DOS、键轨道分析COHP、Bader电荷等)来分解吸附稳定化的来源。
总体而言,DFT以高精度提供了分子筛与分子相互作用的本质认识。例如,Cnudde等人利用静态DFT计算揭示了烯烃与沸石酸中心的π配位作用,说明烯烃在酸性位附近可形成π复合物,从而降低扩散能垒并促进扩散。
又如,通过DFT校正力场参数,Amirjalayer等构建更准确的分子筛-MOF混合力场,成功预测了客体分子在MIL-53呼吸型MOF中的吸附扩散行为,与实验结果吻合。
虽然DFT计算量大、难以直接模拟大尺度体系,但其在关键步骤能垒计算、活性位电子结构表征等方面的作用不可替代,为分子筛催化和吸附机制提供了可靠的理论依据。
分子动力学(MD)模拟
MD通过求解牛顿运动方程模拟原子/分子在给定势能面下的运动,适合研究分子筛体系的动力学过程和统计算律。经典力场MD在分子筛领域应用历史悠久,是解析孔道内扩散和输运性质的主要工具。
基于经验或DFT拟合得到的分子间力场,MD模拟能够在纳米尺度追踪客体分子在分子筛孔道中的运动轨迹,并通过充分采样获得平衡和输运性质。例如,通过MD可以计算客体分子的自扩散系数、扩散活化能,以及观察不同温度、不同负载条件下分子在孔道内的构型演化。
大量研究利用MD系统考察了扩散与多种因素的关系:如温度升高如何加快烷烃在ZSM-5直孔道方向的扩散、客体分子体积和形状如何影响其在微孔/笼状结构中的迁移、分子筛骨架的柔性对扩散是否有促进作用等。
值得一提的是,MD还能揭示混合物竞争吸附和扩散现象,例如模拟显示支链烃在双孔道分子筛中往往被困于交叉孔附近,需克服较高的跳跃能垒方可移动;而在多组分体系中,不同分子之间的相互作用会导致非理想的扩散行为。
近年来,随着计算能力提升,从头算分子动力学(AIMD)也开始用于分子筛研究,在考虑量子效应的同时描绘有限温度下原子运动。例如,有研究通过AIMD计算了乙烯/丙烯在CHA型分子筛中的自由能曲线,发现含硅磷酸盐分子筛较硅铝沸石具有更低的扩散能垒,源于骨架柔性和较大孔窗尺寸的贡献。
总体来说,MD方法以时间和空间尺度上的优势,能够直接模拟气体分子在分子筛孔道中的动态行为,为理解扩散限制对催化和吸附性能的影响提供了直观依据。

DOI:10.11949/0438-1157.20231015
蒙特卡洛(MC)模拟
Monte Carlo方法通过随机采样统计热力学平均,特别适用于模拟吸附平衡和构型分布等问题。在分子筛气体吸附研究中,广泛采用的是巨正则系综蒙特卡洛(GCMC)模拟。
GCMC在给定温度和化学势(或压力)下,通过随机插入、删除和移动分子,不断采样体系的吸附构型,从而计算出平衡吸附量、选择性以及吸附热等热力学性质。这一方法非常适合预测分子筛对单组分及多组分气体的吸附等温线、组分竞争吸附行为等。
例如,在筛选CO₂捕集材料时,研究者常对数千种MOF结构进行GCMC模拟,评估其在特定压力下的CO₂吸附量和CO₂/N₂选择性。通过这种高通量计算筛选,已经从庞大的MOF数据库中发现了一批具有优异气体分离性能的候选材料,并结合数据挖掘揭示了结构–性能之间的关联规律。
除了吸附热力学,蒙特卡洛法还可以用于反应动力学模拟(如KMC,动力学蒙特卡洛),在已知反应机理和速率常数的前提下,随机推进反应事件来仿真催化反应过程。这在复杂网络反应(例如甲烷在分子筛上的逐步转化、MTO中烃池机制)研究中非常有用,可连接DFT计算得到的微观反应速率与宏观的产物分布和TOF(周转频率)预测。
总体而言,MC方法以快速求解平衡和统计问题见长,对于评估分子筛吸附剂的容量与选择性、以及考察催化反应路径的概率分布等方面具有独特优势。在实际研究中,常常将GCMC与MD结合:先用GCMC确定吸附平衡态下气体在孔道中的分布,然后以此构型为初始进行MD,以考察扩散动力学。
这种模拟策略能全面表征分子筛在给定条件下吸附–扩散综合性能,为设计高效吸附分离工艺提供指导。
综上,DFT、MD、MC三种理论模拟手段各自从电子结构、原子运动和统计热力学层面为分子筛研究提供支撑。它们已经成功应用于解释实验观察、预测材料性能并指导新材料开发。
例如,通过模拟可定量比较不同分子筛对CO₂的吸附能力,进而优化吸附剂配方;通过计算反应能垒可以筛选最优催化材料及合理设计助剂改性策略;通过扩散模拟能够揭示孔道尺寸对催化选择性的影响,为分子筛孔结构调控提供依据。
可以说,理论模拟已成为分子筛领域与实验研究并行互补的研究手段,大幅加速了对复杂气体催化与吸附体系的机制理解和应用开发。
总结
分子筛材料凭借独特的微孔结构和表面化学,在气体吸附和催化中展现了不可替代的作用。从传统硅铝酸盐沸石到新兴的MOF、COF框架,研究者不断丰富分子筛家族以满足不同工业需求。
总结而言,分子筛的结构特性(规整孔道、可调酸位等)赋予其对客体分子的择形吸附和限域催化功能,使其在提升目标产物选择性、提高反应速率等方面取得显著效果。
同时,现代理论模拟技术的发展为剖析这些现象的微观机制提供了强有力工具:DFT阐明反应活性与分子筛结构之间的本质联系,MD揭示动态扩散和转化行为,MC则预测平衡吸附和宏观性能。这些手段相结合,已成功解释了许多实验难以直观探测的问题,并指导了新材料和新工艺的设计。
展望未来,分子筛在能源、环境等领域仍将是重要的功能材料,而多尺度模拟与机器学习的引入将进一步拓展研究深度。
例如,借助机器学习构建高精度势能面,可以将DFT级精度应用于更大尺度的MD模拟,加速探索上百万种假想分子筛结构的性能。同时,原位表征技术的发展将与理论计算紧密结合,实现对催化过程的Operando模拟与验证。
可以预见,在实验和模拟交叉印证下,我们将更加深入地理解分子筛孔道内发生的种种奇妙化学,为开发更高效的吸附剂和催化剂提供科学指南,推动气体催化与吸附技术的不断进步。