

电催化二氧化碳还原(CO₂RR)是利用可再生能源将CO₂转化为高值燃料和化学品的关键技术,被视为应对气候变化和能源危机的可持续路径。催化剂表面的电子结构直接决定了反应物与中间体的吸附与活化行为,其中过渡金属催化剂的d电子特征起着主导作用。
早在1990年代,Nørskov等人提出了d带中心理论,表明过渡金属表面d态中心位置与吸附能之间具有直接关联。
相似地,在非金属或主族元素催化体系中,p轨道电子也成为影响催化性能的重要因素:如结合理论和实验发现,特定的p带中心(如氧化物中氧2p、半导体中的主族元素pz轨道等)可以用来描述催化活性。
因此,研究d带中心与p带中心的形成机理及其对CO₂RR性能的影响,对于催化剂的分子层面设计和性能提升具有重要意义。
本文华算科技系统回顾d带与p带中心在不同催化材料(包括金属、金属氧化物、单原子催化剂、二维材料)中的物理来源和形成机制,分析二者的本质差异与相互联系,并结合典型研究和理论模型探讨它们对CO₂RR催化活性和选择性的调控机制,最后总结基于调控d/p带中心的催化剂设计策略和发展趋势。
在过渡金属催化体系中,d带中心(d-bandcenter)是描述催化剂表面电子结构的重要参数,其定义为金属表面d态密度分布的加权平均能量位置,相对于费米能级的高低直接影响吸附键能和反应活性。
当催化剂是金属块体或纳米结构时,由于金属原子间d轨道强烈重叠,形成宽阔的d能带;金属表面和缺陷位点处的d态中心会漂移,反映出金属原子价轨道与吸附物的耦合强度。
根据Nørskov和Hammer的d带中心理论,在多种吸附体系中观察到:d带中心位置越高,催化剂与吸附物之间的反键轨道能级越高,被占据的比例降低,从而导致更强的吸附结合。
相反,当d带中心位置较低(离费米面较深)时,更多反键态被填满,吸附弱化。d带中心可通过密度泛函计算获得,并已被用作机器学习建模金属表面吸附性能的关键特征。

DOI: 10.1016/j.isci.2025.113080
与过渡金属不同,在主要由主族元素构成的催化体系(如金属氧化物、低维碳基材料或纯有机催化剂)中,催化活性往往由p轨道电子主导。在这些体系中可引入p带中心(p-bandcenter)的概念:它通常定义为主族原子(如硼、氧、碳等)特定p轨道态的能量中心。
其物理来源可以是金属氧化物中的氧2p轨道、水合表面上的OH⁻轨道,或是石墨烯、g-C₃N₄等碳基材料中的pz轨道。当催化剂缺乏过渡金属d轨道时,p带中心常被提作替代性的电子结构描述符。例如,在固体氧化物燃料电池阴极材料中,氧2p带中心位置能够影响导电性和氧还原活性;在非金属单原子或二维催化剂中,掺杂原子的pz能级位置同样调控吸附性能。
总之,d带中心反映了过渡金属催化剂中d电子的平均能级,而p带中心则描述了以p轨道为主导的体系中的电子能级特征,它们共同构成了电子结构的核心指标。
具体材料中,d带和p带的形成机制又各有差异:对于金属和合金纳米催化剂,d带源于金属原子价d轨道在周期性晶格中的重叠,带中心位置受金属种类、晶面、应变和邻近原子配位影响。如在单晶Pd和Cu不同晶面间,本构金属的d轨道重叠差异决定了d带中心的微小位移。
在金属氧化物和碳化物中,除了金属d带外,氧或碳的p轨道与金属轨道混成,形成复合的价带和导带结构。如钙钛矿LaCoO₃中,不同A位掺杂可以将费米能级向氧2p带推移,减少金属3d与氧2p之间的能隙,从而增强Co–O键的共价性。
对于单原子催化剂(SACs)与双原子位点结构,孤立的过渡金属原子提供局域的d轨道状态,其能级受邻近配体(如N、C或O)p轨道杂化而改变。例如,Fe–N₄基催化剂中Fe的d带中心受配位氮原子场的调制,显著影响CO₂中间体的吸附特性。
二维催化材料则更为多样:典型的2D过渡金属硫化物(如MoS₂)具有金属M的d带和硫X的p带,界面和缺陷可以改变二者的交互;而纯碳二维材料(如石墨烯、g-C₃N₄)和硼氮烯等,主要靠p轨道(如C/N的pz)贡献导电带。
综上所述,d带中心和p带中心虽来源不同,但均反映了催化剂局部原子电子能级的重组规律,是理解催化活性本质的关键。
核心区别与联系
从电子结构角度分析,d带中心与p带中心表现出本质差异。首先,过渡金属的d轨道具有较强的局域性和方向性,它们在催化表面形成的d带通常较窄。更重要的是,d轨道通常位于费米能级附近,参与金属–吸附物键的形成和反键结构的分布。
因此,上移(接近费米面)的d带中心意味着反键态未完全填满,从而增强了吸附物与金属的结合强度;而下移的d带中心则相反,吸附强度减弱。
相比之下,p带中心对应的p轨道(如元素的pz或氧的2p)在能级上往往更低且较为离散,其能带宽度也较大、延展性更强。在没有d轨道的催化体系中,p轨道与费米能级的耦合关系决定了吸附键的极性与能量。
如氧化物催化剂中,当氧的p带中心向费米能级靠近时,金属–氧键的共价性增大;这意味着更易发生电子转移和键裂解过程。
DOI: 10.1016/j.isci.2025.113080
此外,d带中心与p带中心虽不同,却可以协同影响催化反应。许多催化材料中存在d-p轨道耦合:例如在过渡金属氧化物或混合金属结构中,金属d轨道和近邻原子的p轨道杂化程度是决定表面电子结构的关键。
文献报道,改变结构对称性或引入杂化原子可以强化d-p偶联。在一项典型研究中,研究者在Zn-Sn双原子位点中部分掺杂S原子后,打破了位点的局域对称性,显著增强了Sn原子的p轨道与Zn原子的d轨道之间的耦合,使得*HCOO中间体更稳定地吸附并降低反应能垒,最终大幅提升了CO₂还原生成甲酸盐的选择性。
这表明在设计催化剂时,调控p轨道和d轨道的相对能级与耦合强度是一种有效策略。类似地,在过渡金属–氧化物混合界面上,将费米能级调整到靠近氧的2p带中心,可以提高金属–氧的共价性,使含氧中间体更易形成。因此,d带中心和p带中心虽然源自不同轨道,但在复合体系中往往相互联动,共同决定吸附和反应性质。
综合来看,d带中心与p带中心的区别在于它们源自不同类型的原子轨道(过渡金属d轨道 vs. 主族元素或氧的p轨道)以及不同的能级特性(通常d带更靠近费米面,p带偏低)。
但它们也具有共通点:都可作为催化剂表面电子能级的指标,影响吸附能和活性。而二者的相互作用(例如过渡金属中心与周围非金属配体的d–p杂化)则是决定电子分布和化学键性质的重要因素。
对催化性能调控的机制
在CO₂RR中,催化剂必须促进CO₂分子的吸附、活化以及中间体向目标产物的转化。d带中心理论为这种机理研究提供了有力工具:一般认为,适中的d带中心位置可以使催化剂与关键吸附物(如COOH、CO、HCOO 等)的键合强度达到最优平衡,从而提高催化活性。
例如,某些研究显示,铜表面上的不同晶面因d带中心位置不同而导致催化活性差异:Cu(100)面上d带中心更接近费米能级,与CO的π轨道重叠更大,从而对CO吸附更强,最终在CO₂电还原中展现出更高的C₂+产物选择性。
同样的机理也解释了为何掺杂可以提高金属催化剂性能:研究者发现,引入拉伸应变或表面拉伸应力能上移过渡金属纳米结构(如Pd纳米孪晶)的d带中心,显著增强表面对CO₂分子的吸附和活化,极大地提高了CO₂还原生成CO的效率。这里的核心是:上移的d带中心降低了吸附反应的能垒,使得反应在较低过电位下进行。
对于基于p轨道的催化剂,p带中心的位置同样决定中间体的吸附强度和反应路径。以p区元素催化剂为例,Sn、In、Bi等金属或氧化物基催化剂广泛用于将CO₂还原为甲酸盐。
在这类体系中,理论和实验都表明,较高的p带中心(能级更接近费米面)会加剧吸附物之间的电子排斥,减弱HCOO中间体与催化位点的键合;反之,将p带中心下移可以提高HCOO的结合力。
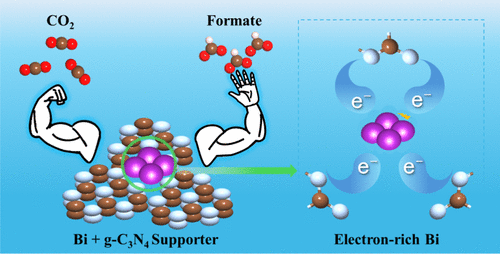
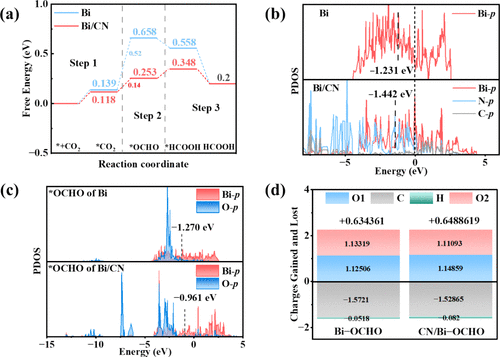
例如,王等人报道,通过设计Bi纳米片与g-C₃N₄复合结构,非金属g-C₃N₄向Bi位点输送电子,使Bi原子的p带中心下移(相对于费米能级更负),并伴随Bi与OCHO中间体之间电子转移增加,从而显著增强了OCHO吸附和甲酸的产率。
这一结果说明了:调制p带中心同样可以优化CO₂RR的催化路径和选择性。换言之,如果p带中心下移(更深的p能级),则吸附态电子更丰富,*OCHO等含氧中间体得到强化吸附,有利于向甲酸盐方向的转化。
DOI: 10.1021/acsami.5c03240
此外,不同催化路径中酸碱理论(Lewis酸碱)也是性能调控的重要视角。CO₂分子本身略显路易斯酸性,其碳原子电子负性不强,因而易与具有路易斯碱性的位点(如O²⁻、N原子或疏电子的掺杂原子)发生相互作用。
在一些单原子或双原子催化剂设计中,人为制造出邻近的路易斯酸碱对位点,可以显著促进CO₂的活化。例如,通过向金属位点掺杂B、N等主族元素,可以形成新的酸性中心或碱性中心,从而改变CO₂分子在催化面的取向和吸附方式,进而影响反应途径。
虽然相关实例较多,但其本质也是通过调控电子结构(带中心)来改变吸附景观。最后,异质界面或界面电子态耦合效应也不可忽视:不同材料界面往往会出现新的电子态,如掺杂金属/氧化物界面或碳/金属界面可以形成金属–非金属混合轨道,使得界面处的d带和p带发生重组,进而促进中间体的快速转化。
例如,双功能催化剂表明,当金属d带与邻近氧物质的p带耦合加强时,可以实现CO₂RR的吸附和脱附动态平衡优化,提高电催化活性。综合以上理论与实例可见,d带中心调控与p带中心调控是两种相辅相成的机理视角:前者适用于描述过渡金属位点与吸附物之间的键能关系;后者则适合非金属或氧化物体系,描述间接的电荷转移和极性相互作用。
在CO₂RR的具体反应路径(如COOH生成CO或C1+产品,*OCHO生成HCOOH等)中,催化剂的d/p带位置决定了哪个路径的中间体更易稳定,从而影响最终产物分布。
催化剂设计的应用策略
基于对d带与p带中心机理的理解,近年来研究者提出了多种提高CO₂RR催化性能的设计策略,其核心均围绕着有针对性地调控催化位点的电子结构。一类重要手段是掺杂与合金化:通过引入异质元素改变原位点的电子态。
例如在过渡金属催化剂中,掺入电子性能不同的金属或非金属原子,可有效移动d带中心位置。文献报道,在Cu、Pd等金属纳米结构中掺杂异原子不仅可以改变自身d带中心,还可能改变邻近位点的化学性质。
在以Sn和Bi为代表的p区金属催化剂设计中,通过掺杂S、N或O等非金属原子,可以显著优化p带中心及配位环境,从而增强对HCOO的吸附。例如,北京理工大学陈文星课题组通过在Zn–Sn双原子位点中引入部分S掺杂,成功调节了Zn中心的电子结构并强化了Zn–Sn双位点之间的p–d耦合,显著稳定了HCOO中间体,最终使CO₂还原产甲酸效率大幅提高。
类似地,在Bi基催化剂中,g-C₃N₄等非金属支持不仅提供额外电子,亦通过界面电荷传递使Bi的p带中心下移,成功提升了*OCHO吸附强度并提高产率。这些例子表明:精确调控掺杂位点或复合界面能够系统地调节d/p带中心位置,从而优化催化反应路径。
DOI: 10.1021/acsami.5c03240
另一种策略是应变与形貌工程:施加拉伸或压缩应变可以显著改变催化剂表面原子的电子能级结构。如前述Pd纳米孪晶研究表明,拉伸应力使Pd的d带中心上移,强化了对CO₂的吸附和活化。
类似地,对于二维材料和柔性催化剂,应变调控(如弯曲、压应力)能够调整d/p能带结构,进而影响吸附态。例如,对MoS₂单层施加应变后,其边缘Mo的d带中心和S的p带中心会发生可控移动,从而改变CO₂中间体的结合形式。此类应变策略通常通过设计核壳结构、层间界面或选取合适的基底来实现。
此外,缺陷工程和界面设计同样是调控d/p带中心的重要途径。制造氧空位、硫空位等缺陷可以诱导局部电子重分布,常常导致附近金属d带或缺陷处p带上移。例如,在Bi₂O₂CO₃中引入氧空位,可以升高氧的p带中心,使CO₂中间体(如*OCHO)更易吸附。
异质界面(如金属/氧化物、金属/碳复合物)则通过界面电子态重组形成新的能级分布。设计具有适中氧化态或表面组装的复合催化剂,可以获得理想的d/p能级对,如某研究显示Bi/Bi₂O₂CO₃界面上Bi的p带中心介于金属Bi与氧化态之间,使催化体系同时兼具电子丰富性和中等吸附强度,实现了高选择性还原。
总之,通过掺杂调控、应变调节、缺陷设计以及多相界面工程等手段,可以精细调节催化剂的d带中心或p带中心位置,从而在分子层面上优化CO₂RR的活性与选择性。
总结
综上所述,d带中心和p带中心代表了过渡金属催化剂和主族元素催化剂电子结构的两种典型标志,它们分别反映了不同类型原子轨道与吸附物相互作用的强度和特征。
在电催化CO₂还原中,合理调控d带中心位置能够改变催化剂对碳反应中间体的吸附能,从而优化关键反应步的能垒;而调节p带中心则在主族金属或非金属催化剂中发挥类似的作用,特别是对含氧中间体的吸附有显著影响。
现有研究已通过理论计算和实验证实,提升d带中心(向费米面靠近)往往加强对吸附物的结合,而降低p带中心(能级更深)有利于增强对某些极化中间体的稳定。基于此,掺杂异构元素、施加应变、制作界面和缺陷等策略已被广泛应用于新型CO₂RR催化剂设计中,并取得了一系列性能突破。
未来,深入探索d带中心与p带中心之间的耦合效应有望进一步拓宽催化剂优化思路。例如,双功能或多功能位点催化剂(兼具高d带和可调p带)可能实现CO₂分子向不同产物路径的协同控制。
此外,将第一性原理计算与机器学习相结合,以高级描述符定量捕捉d/p带中心变化对多电子过程的影响,也是提高催化剂设计效率的方向。总之,理解d带中心与p带中心在纳米尺度下的结构化控制规律,将为开发高效、稳定且选择性可控的CO₂电还原催化剂提供指导。
随着催化材料学科的发展,我们有望构建出更具原子级精度的电子结构调控平台,进一步实现CO₂到碳基燃料与化学品的绿色高效转化,为能源与环境问题的解决贡献力量。