

说明:本文华算科技介绍了晶格氧与氧空位的概念、晶格氧参与反应的机理(包括热催化氧化的MVK机理、电化学晶格氧介导机制以及高级氧化中晶格氧活化的机制)、晶格氧活性调控策略(非活性A位阳离子取代和催化剂后处理)。
晶格氧:构成晶体结构的体相氧被称为晶格氧,在热氧化催化和电池正极材料设计应用中经常被提及。
氧空位:是指在金属氧化物或者其他含氧化合物中,晶格中的氧离子(氧离子)脱离,导致氧缺失,形成的空位。

图1. 氧空位示意图。DOI: 10.12677/APP.2023.138039
MVK是一种两步氧化还原反应模型,常用于解释过渡金属氧化物催化剂的氧化反应速率。在气相催化反应中,MVK机制涉及晶格氧参与反应的过程。挥发性有机化合物(VOCs)首先与催化剂中的晶格氧反应,生成CO2、CO和H2O(g),随后通过B(n+1)+位点到Bn+位点的过程激活O₂分子以生成活性氧,如图2所示。
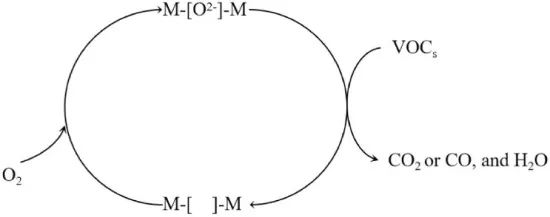
图2. Mars-van Krevelen机制示意图。DOI: 10.1016/j.apcata.2023.119348
MVK模型将VOCs氧化过程分为两步:第一步,反应物与催化剂中的晶格氧发生初始反应,消耗晶格氧并产生氧空位,导致催化剂被还原;第二步,氧填补空位,使晶格氧再生,催化剂被再氧化。
MVK机理涉及催化剂先被还原,随后又被氧化,因此也被称为氧化还原机制。在稳态操作中,还原和氧化步骤的速率必须相等。该模型已被广泛应用于金属氧化物催化剂上的烃类完全氧化过程。
有研究表明,催化剂中的晶格氧可以被激活,伴随催化剂中较高价态金属的还原,形成活性氧和氧空位,触发活性氧物种(ROS)的连续形成,如羟基自由基(•OH)、超氧阴离子(O₂⁻)、单线态氧(¹O₂)等,如图3所示。
晶格氧被视为氧化过程中催化性能的良好指标。相关研究表明,晶格氧含量越高的催化剂,其活性越高。
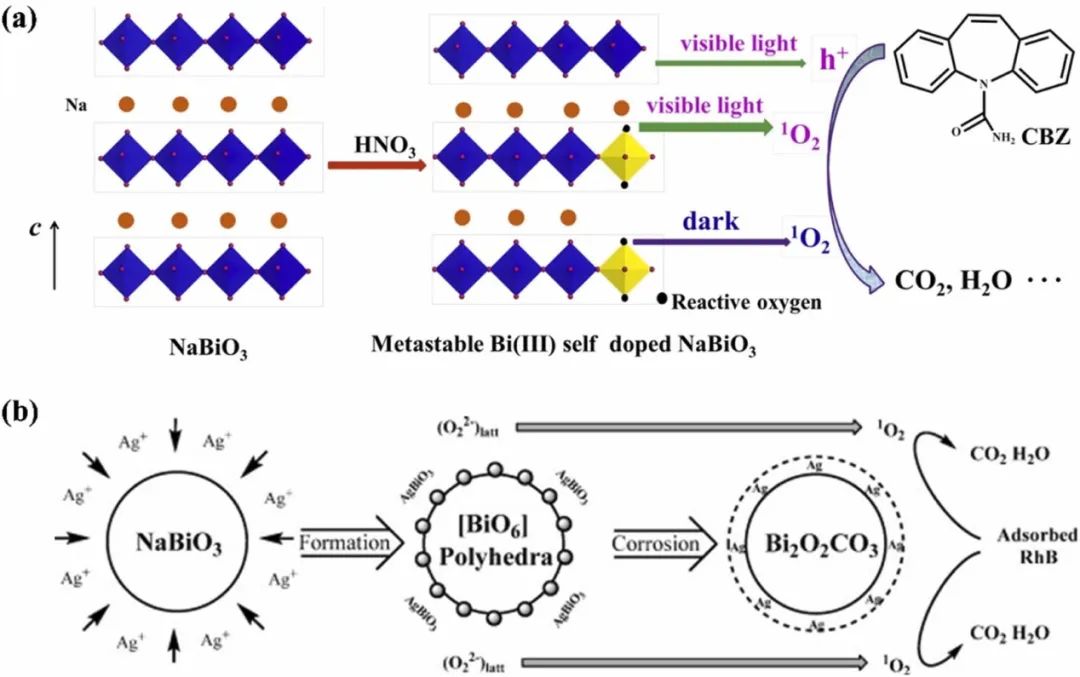
图3. (a)亚稳态NaBiO3(NBO)纳米片上的光腐蚀反应增加了NBO晶格氧中单线态氧的生成。(b)生成单线态氧作为主要活性物种的Bi₂O₃-Ag₂O(BSO)的形成及其对罗丹明B(RhB)染料的降解。DOI: 10.1016/j.apcatb.2016.09.054
电化学晶格氧介导机理(LOM)是电催化析氧反应(OER)的一种重要机制,其核心是晶格氧通过自身氧化参与水氧化过程。
LOM不同于传统的吸附质演化机理(AEM),可加速晶格氧的直接耦合,最小化反应能垒,为提高OER效率提供新思路。该机理中,晶格氧通过自身氧化形成氧分子参与水氧化,需将氧的2p能带向上移动以接近费米能级,增加其与金属d带的轨道重叠(即增强M-O键共价性),使晶格氧的氧化还原在能量上更易进行。
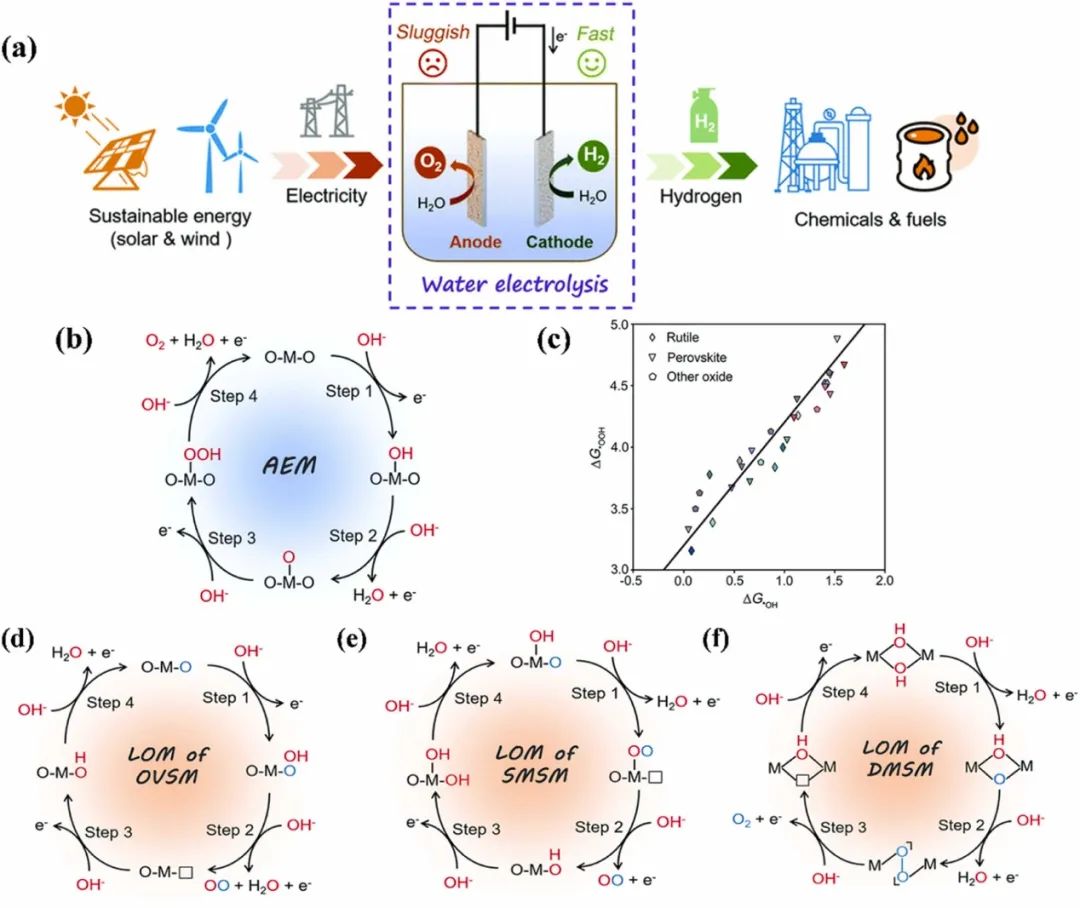
图4. (a)通过水电解在有价值的化学品和燃料中储存大量能量的示意图。(b)在碱性介质中活性金属位点上析氧反应(OER)的吸附–解吸机制(AEM)示意图。(c)各种过渡金属氧化物*OOH和*OH结合能之间的标量关系。(d)氧空位位点机制(OVSM)。(e)单金属位点机制(SMSM)和(f)双金属位点机制(DMSM)。DOI: 10.1038/nmat4551
根据活性中心的不同,LOM的OER路径可分为三类:
氧空位位点机制(OVSM)中,活化的晶格氧作为活性位点接受OH⁻形成*OOH物种,释放O2后产生的氧空位再被OH⁻填补;
单金属位点机制(SMSM)中,OH⁻在单个金属位点吸附后发生去质子化,*O中间体与活性晶格氧直接耦合;
双金属位点机制(DMSM)中,相邻活化晶格氧通过分子内氧耦合形成M-OO-M基序。
晶格氧活化是LOM发生的前提,要求催化剂具备独特的电子结构,尤其是晶格氧自身的电子特性。
在高级氧化过程(AOPs)中,晶格氧的活化机理主要遵循MVK机理,具体如下:高级氧化过程通过高氧化能力的活性氧物种(ROS,如羟基自由基·OH、硫酸根自由基SO₄⁻·、单线态氧¹O₂等),将有机物氧化为H2O和CO2,其中涉及晶格氧的活化主要与黑暗环境下的热催化相关,该过程遵循MVK机理。
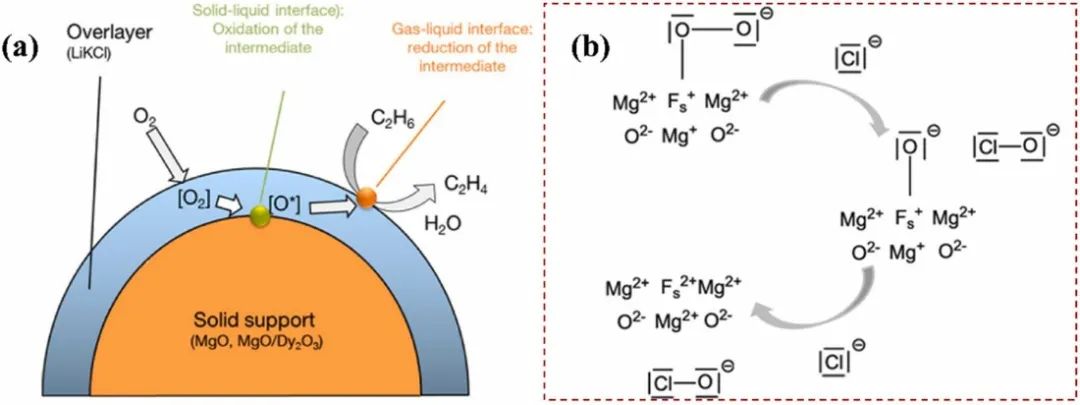
图5.(a)乙烷氧化脱氢(ODH)在负载型碱金属氯化物催化剂上的反应序列示意图。(b)氯化物在氧化镁氧空位上协助O₂活化的可能分子路径。DOI: 10.1021/ja505411s
黑暗环境条件下热催化中晶格氧的MVK机理分为两步:第一阶段是催化剂表面晶格氧迁移率提高,晶格氧氧化杂质并使部分催化剂还原;随后,被还原的催化剂被表面吸附的氧再氧化。催化剂内的晶格氧被活化形成活性氧物种(O*)和氧空位,进一步促进降解过程中ROS(如1O2、O2-和·OH)的形成(步骤2)。
因此,氧空位数量增加,作为氧吸附的活性位点,进而再次形成晶格氧。该过程中的速率决定阶段被认为是催化剂与污染物之间的氧转移。MVK过程中还原态催化剂的再氧化对催化剂的稳定性至关重要。
非活性A位阳离子取代是调控晶格氧活性的重要策略之一,主要通过在催化剂的A位引入其他阳离子,改变材料的结构与电子特性,从而影响晶格氧活性。
在钙钛矿氧化物(结构通式ABO3)中,A位通常为稀土或碱土金属,通过部分或完全取代A位阳离子(如用低价Sr2+取代La3+),可调节晶格缺陷、B位阳离子价态及氧空位浓度。
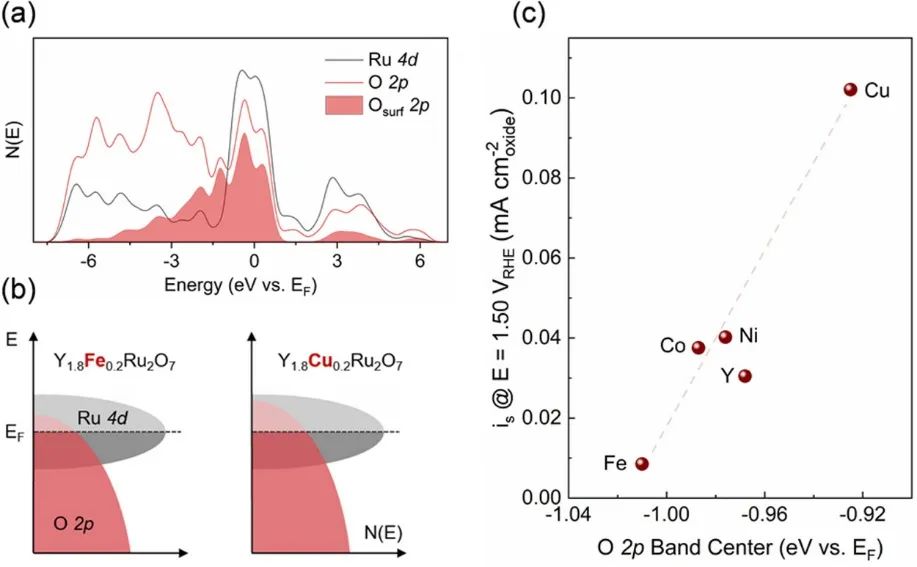
图6. (a)Y1.8Cu0.2Ru2O7中Ru 4d和O 2p态的投影态密度(DOS)。表面终止氧原子的O 2p态的投影DOS被阴影标记。(b)Y1.8Fe0.2Ru2O7和Y1.8Cu0.2Ru2O7的示意图刚性带图,展示了O 2p带中心的上移,这有助于晶格氧的损失、氧空位的形成以及氧析出反应(OER)活性的增强。(c))Y1.8M0.2Ru2O7−δ的特定OER活性(以1.50 VRHE时的电流密度估算)与O 2p带中心位置之间的相关性。DOI: 10.1021/jacs.0c01135
在烧绿石钌酸盐氧化物(结构通式A2B2O7-δ)中,A位取代(如Y1.8M0.2Ru2O7-δ中M=Cu、Co等)可控制表面氧空位浓度。例如,Cu取代A位使费米能级接近O 2p能带中心,增强Ru-O键共价性,提升氧空位浓度和OER活性;Sr²⁺取代Y³⁺可改善配位几何,减弱电子关联,加快电荷转移,降低OER反应能垒。
非活性A位阳离子取代通过调控氧空位浓度、金属–氧键共价性及电子结构,有效增强晶格氧活性和催化性能。
催化剂中氧空位的数量影响晶格氧的活性。通常,氧空位会随着阳离子缺陷的产生而自发出现,以维持电荷中性。这种缺陷特征有利于晶格氧的迁移、释放和嵌入,可采用后处理方法控制氧空位的浓度。
① 气相热处理
气相热处理是调控催化剂氧空位浓度、增强晶格氧活性的一种后处理方法。气相热处理通过在缺氧环境(如H2、CO、N2、Ar、真空等)中适当温度下退火,促使金属氧化物表面或体相释放晶格氧,形成氧空位。
其原理为晶格氧在金属氧化物作用下转化为氧空位,并释放O2和电子,氧空位浓度与氧分压成反比,缺氧条件更利于其形成。
例如,研究人员通过在氢气气氛中800℃退火,将立方钙钛矿氧化物BaZr0.5In(III)0.5O2.75转化为高度阴离子缺陷的羟基氧化物BaZr0.5In(II)0.5O2.25H0.5。
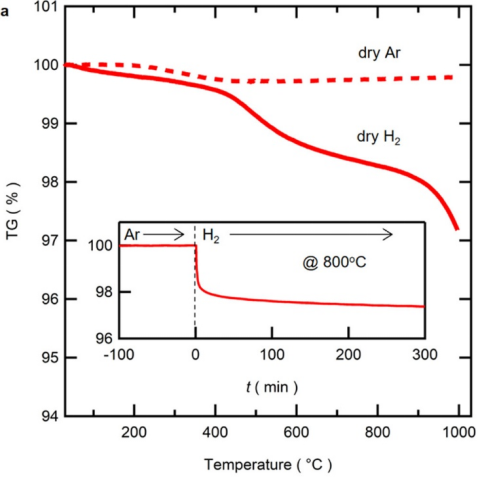
图7. BZI的氢化反应。在干燥的氩气(虚线)和氢气(实线)中测量的热重(TG)曲线。插图:在800°C时,通过将气氛从氩气切换为氢气获得的等温TG曲线。DOI: 10.1021/acs.chemmater.2c01467
② 固相还原
许多固体粉末也被用作还原剂,通过消耗金属氧化物表面的晶格氧原子,导致表面氧空位的形成。例如,研究人员采用球磨辅助固相反应法制备了缺氧Li4Ti5O12-x负极粉末。氧化石墨烯促进Li4Ti5O12-x材料中Ti(IV)向Ti(II)的转化,同时形成氧空位。
③ 溶液反应
溶液反应技术作为高温处理方法的替代,可在温和条件下为金属氧化物引入氧空位。其中,使用NaBH4作为还原剂是最成功且常用的方式,例如研究人员利用NaBH4制备出具有大比表面积和丰富氧空位的铁钴氧化物纳米片,氧空位可增强电子导电性并提供更多催化活性位点。
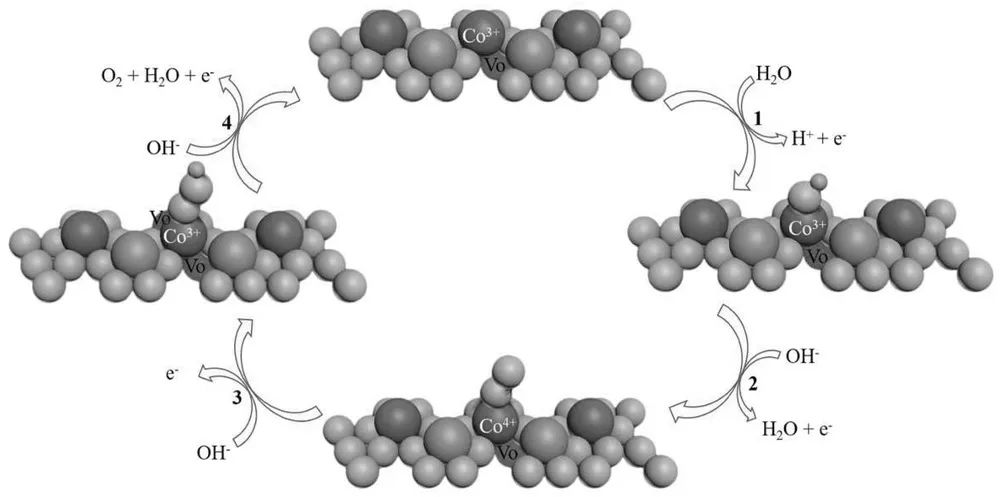
图8. 靠近氧空位的Co3+活性位点的氧析出反应(OER)机制。DOI: 10.1002/adma.201606793
④ 电化学方法
是调控催化剂中氧空位浓度的后处理手段之一。
电化学阳极氧化:在特定电化学环境中,金属基体表面发生氧化反应,生成的金属离子与电解质中的氧反应形成均匀氧化层,可一定程度控制氧空位浓度。
电化学还原:通过阴极还原在金属氧化物中引入氧空位,该方法简单、快速且可扩展性强。例如,采用阴极还原法对TiO2纳米管阵列进行改性,可提高其电化学活性、电导率及超级电容器电容,相比传统热处理大幅降低仪器和资源需求。