

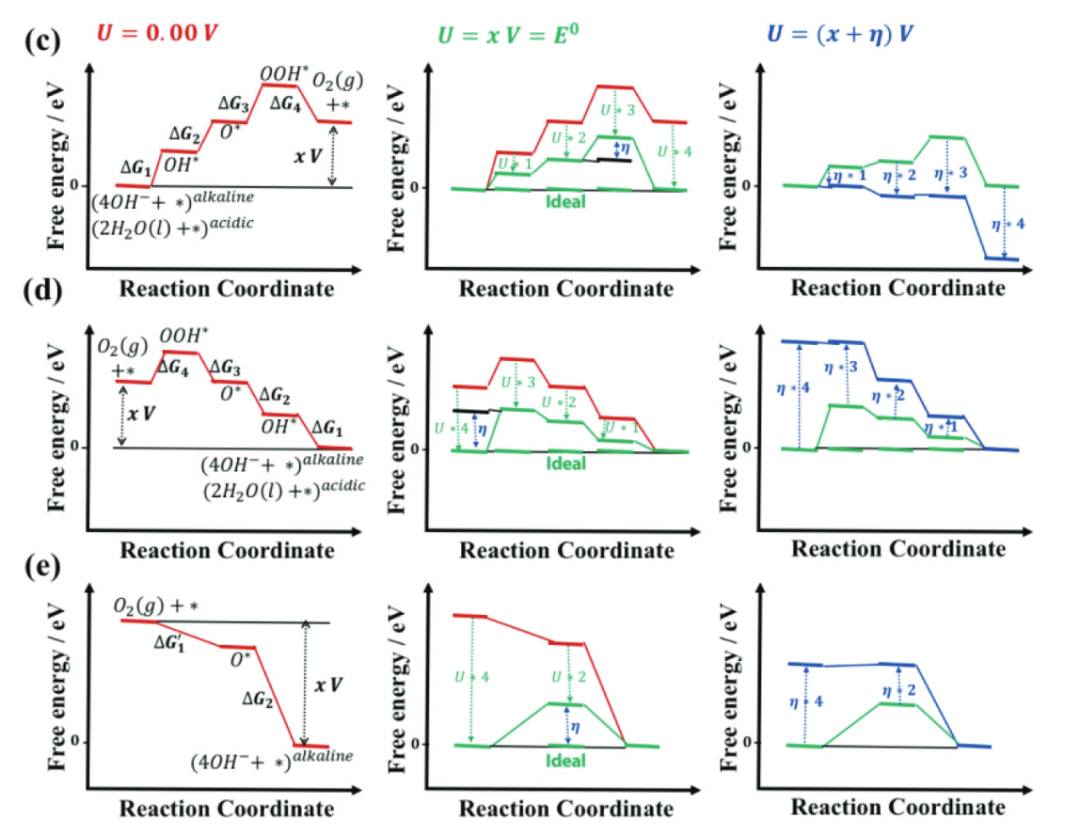
说明:过电位是实际电极电位与热力学平衡电位的差值,量化反应能垒,值越大催化效率越低。其计算依托DFT,通过构建自由能图确定速率决定步骤(RDS)的最大自由能变(ΔG_max),按反应类型公式计算。
应用于OER、HER等电催化反应,经典案例如铁电材料极化调控过电位,为催化剂设计提供依据,推动燃料电池等领域发展。


什么是过电位?

过电位(η)是电催化反应中实际所需电极电位与热力学平衡电位(U₀)的差值,即η=|U_实际-U₀|,其核心物理意义是量化反应能垒——过电位越大,表明反应克服能垒所需的额外能量越高,催化效率越低。
DOI:10.5772/intechopen.77109
在理论计算中,过电位的确定依赖于反应自由能图的构建:通过计算反应路径中各基元步骤的吉布斯自由能变(ΔG),识别出自由能变最大的步骤,该步骤的ΔG_max直接决定过电位的大小。
对于多步反应,过电位的计算公式与反应类型相关:OER的过电位η=(ΔG_max /e)-1.23 V,HER的过电位则为η=|ΔG_max /e|。
过电位的物理本质源于催化剂表面中间体吸附与脱附能垒的非理想性,根据萨巴蒂尔原理,理想催化剂需使各中间体的吸附能处于“适中”范围,偏离这一范围会导致某一步骤的能垒升高,进而增大过电位。
例如,在OER中,关键中间体*OH、*O、*OOH的最优吸附能存在特定关系,若催化剂对OOH的吸附过,则O→OOH步骤会成为RDS,导致ΔG_max增大;若对OH吸附过强,则OH→O步骤的能垒升高,同样会增大过电位。
这种中间体吸附能与过电位的关联,使得过电位成为连接微观电子结构与宏观催化活性的核心描述符——通过调控催化剂的电子态优化中间体吸附能,可有效降低过电位,提升催化性能。
DOI:10.1021/acscatal.1c03737
计算方法与工具
过电位的理论计算依托多层次的方法体系与专业化工具,从电子结构解析到宏观动力学模拟,实现了对不同复杂程度电催化体系的精准描述。
基于密度泛函理论的标准流程是最基础且应用最广泛的方法,其核心步骤包括:首先通过VASP、Quantum ESPRESSO等软件对催化剂表面构型进行结构优化,获得能量最低的稳定结构,确保后续吸附能计算的准确性;随后计算反应中间体在表面的吸附能,这一步需考虑吸附位点的差异,通过对比不同位点的吸附能确定最稳定吸附模式。
接着进行自由能校正,引入零点振动能、熵变及溶剂化效应,将DFT计算的内能变转换为吉布斯自由能变;再通过构建自由能图,绘制各基元步骤的ΔG随反应坐标的变化,直观识别出自由能变最大的速率决定步骤(RDS);最后根据RDS的ΔG_max,按对应反应类型的公式计算过电位。
该流程的关键模型包括计算氢电极和标度关系:计算氢电极通过将电子能量参考系转换为标准氢电极电位,建立ΔG与电极电位的直接关联,使理论计算可模拟不同电势下的反应行为;标度关系则揭示不同中间体吸附能的线性关联,简化了火山图分析,可通过单一描述符预测过电位,大幅减少计算量。
DOI:10.1021/acscatal.1c03737
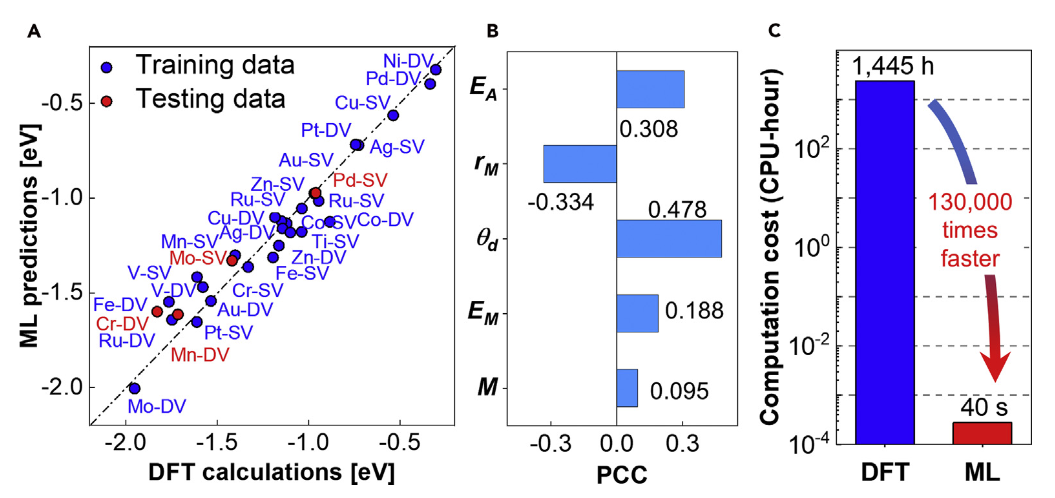
进阶方法与工具进一步拓展了过电位计算的适用范围与效率。机器学习加速通过设计原子描述符训练预测模型,可快速估算过电位,误差控制在7%以内,计算效率较DFT提升10⁵倍以上,例如在单原子催化剂的OER过电位预测中,模型可在数小时内完成数千种组合的筛选,识别出Co/MoS₂(η=162 mV)和Ni@D-Gr(η=197 mV)等高效体系。
混合电位理论适用于多反应路径竞争的体系,可量化不同路径对总过电位的贡献,如在CO₂还原与HER的竞争反应中,该理论能明确过电位对产物选择性的影响。
非DFT模型包括界面碰撞模型和浓度过电位模型:界面碰撞模型模拟质子交换膜燃料电池(PEFC)中质子非弹性碰撞导致的能量损失,通过η=能量损耗/质子通量关联微观碰撞与宏观过电位;浓度过电位模型基于Butler-Volmer方程,量化质量传输阻力,适用于分析传质对过电位的贡献。
DOI:10.1016/j.isci.2021102398
常用软件与数据库为过电位计算提供了技术支撑:DFT计算平台负责电子结构与吸附能求解,支持周期性体系与复杂界面模拟;溶剂化模型通过隐式处理溶剂效应,修正吸附能计算误差;机器学习框架用于描述符设计与过电位预测模型训练;吸附能数据库存储大量预处理的吸附能数据,为标度关系构建与火山图分析提供基础。
这些方法与工具的协同应用,覆盖了从原子级模拟到宏观性能预测的全链条,为过电位的精准计算与催化剂设计提供了全方位支持。
铁电极化调控OER过电位
Lan等人通过系统的理论计算揭示了铁电材料InSnO₂N的极化方向对氧析出反应(OER)过电位的调控机制,为设计低过电位催化剂提供了基于电极化的新策略,其计算流程与结果分析展现了过电位理论研究的典型范式。
DOI:10.1021/acscatal.1c03737
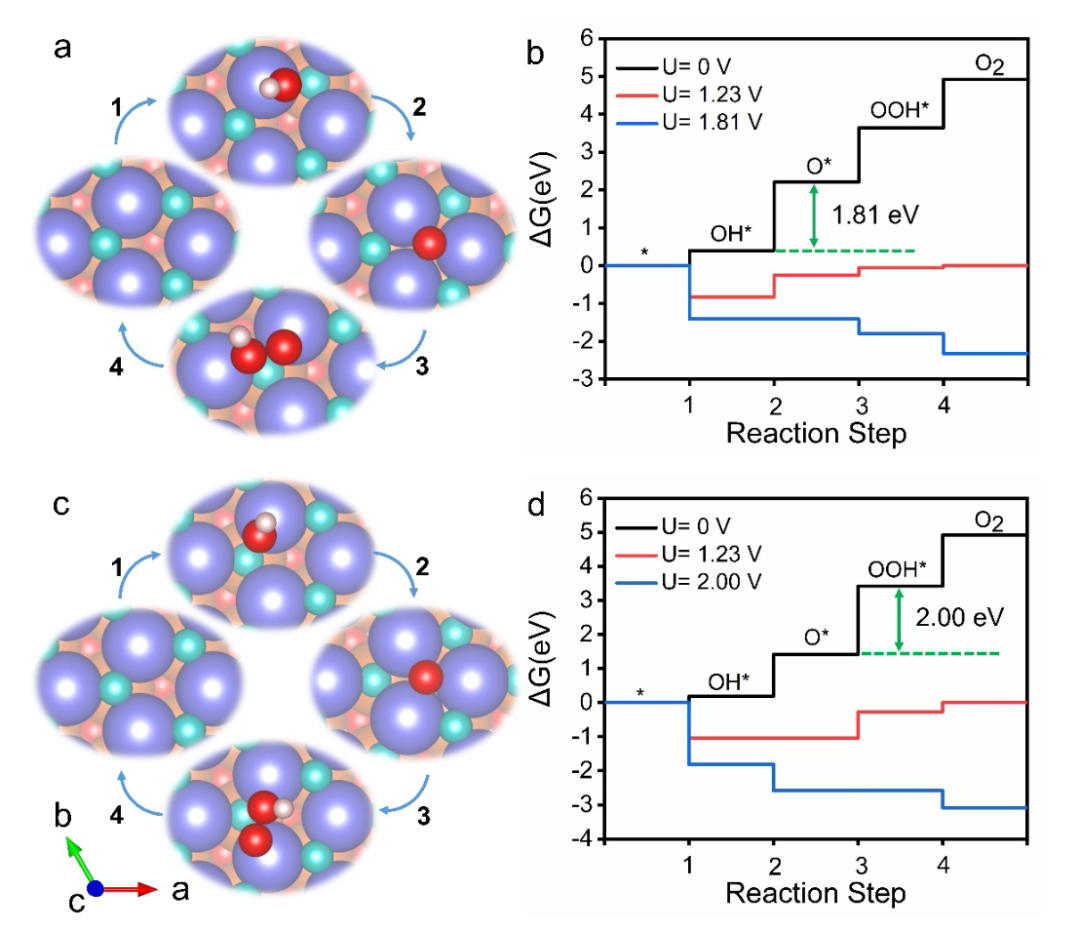
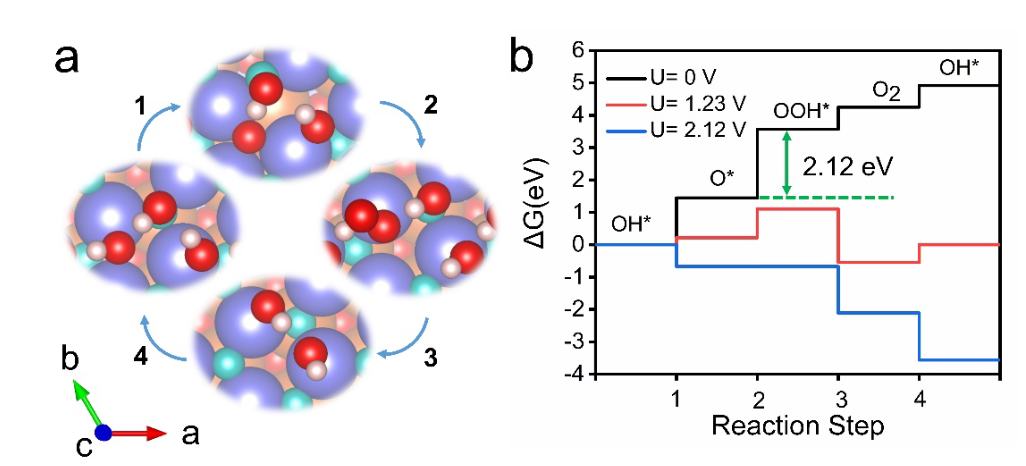
计算目标聚焦于铁电材料独特的极化特性——通过外场调控可翻转极化方向(±P),探究这种极化切换如何影响表面活性位点的电子态,进而改变OER过电位。
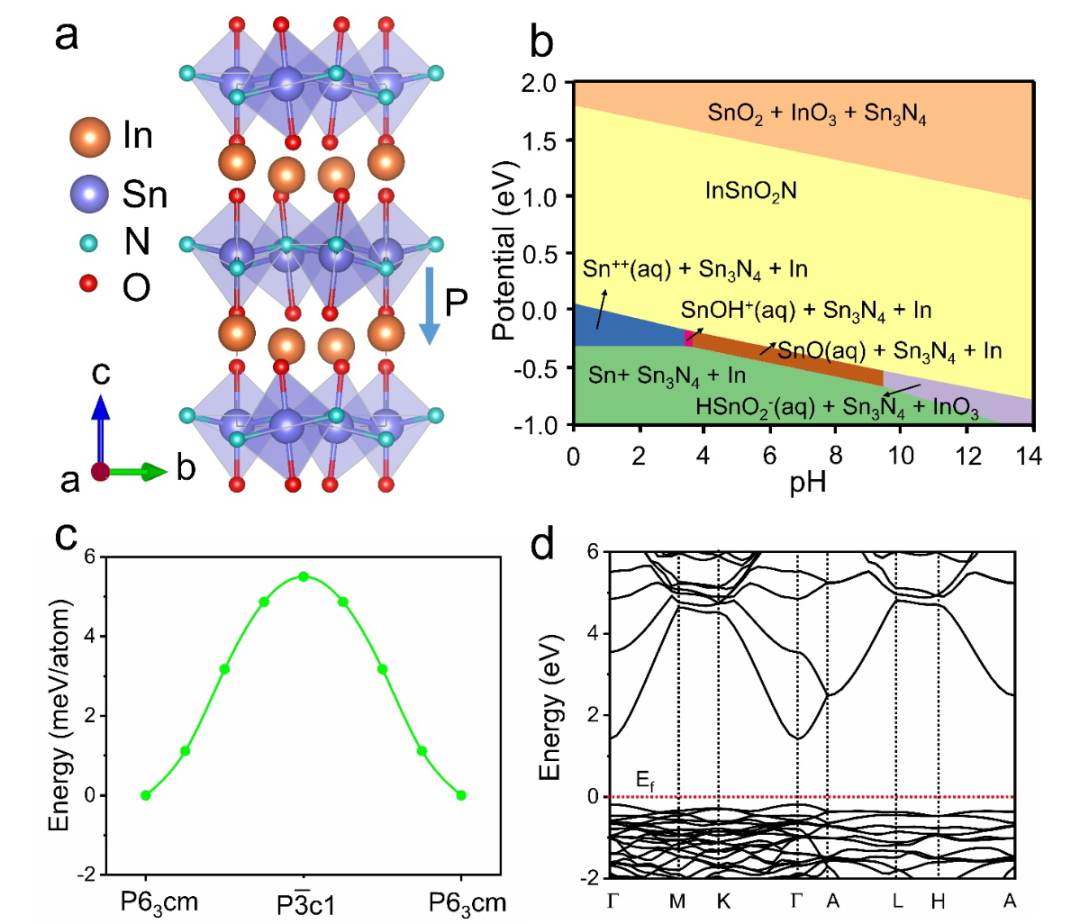
计算流程始于精准的表面建模,构建了InSnO₂N的SnN终止面模型,通过施加正(+P)、负(-P)极化电压模拟不同极化方向,优化后得到稳定的表面结构,其中负极化(-P)使表面Sn原子的配位数从6降至5,形成低配位活性位点。
自由能计算针对OER的四步反应路径展开,通过DFT计算各步骤的ΔE,结合零点振动能、熵变及隐式溶剂化校正,获得各步骤的吉布斯自由能变(ΔG)。
结果显示,不同极化方向下的速率决定步骤(RDS)与过电位存在显著差异:负极化(-P)时,RDS为O→OOH步骤,ΔG_max=1.81 eV,对应过电位0.58 V;正极化(+P)时,RDS变为OH→*O步骤,ΔG_max=2.00 eV,过电位升至0.77 V,表明负极化可使过电位降低0.19 V,显著提升OER活性。
电子结构分析揭示了极化调控的微观机制:电荷密度差分图显示,负极化增强了Sn活性位点的电子密度,这种电子富集使Sn的5s轨道与OOH的2p轨道杂化增强,优化了OOH的吸附能,降低了O→OOH步骤的能垒;而正极化导致Sn位点电子缺失,与OH的相互作用过强,使OH→O步骤成为瓶颈。
表面Pourbaix图进一步证实,在OER工作电位下,负极化表面的OOH覆盖率显著高于正极化,表明负极化更有利于RDS的进行。
该研究的理论意义在于,首次证实铁电极化可通过动态调控活性位点电子态,突破OER中OH与OOH吸附能的标度关系限制——传统催化剂因ΔG_*OOH≈ ΔG_*OH+3.2 eV,难以同时优化两个中间体的吸附能,而过极化调控可打破这种线性关联,使ΔG_*OOH降低的同时不显著影响ΔG_*OH,为设计低过电位OER催化剂提供了全新思路,其方法学也为其他铁电材料在电催化中的应用提供了可借鉴的计算范式。
DOI:10.1021/acscatal.1c03737
典型应用
过电位的理论计算在电催化研究中具有广泛应用,通过自由能图分析与理论-实验关联验证,为催化剂活性评估、机制解析及性能优化提供了量化依据,两个典型应用清晰展现了其价值。
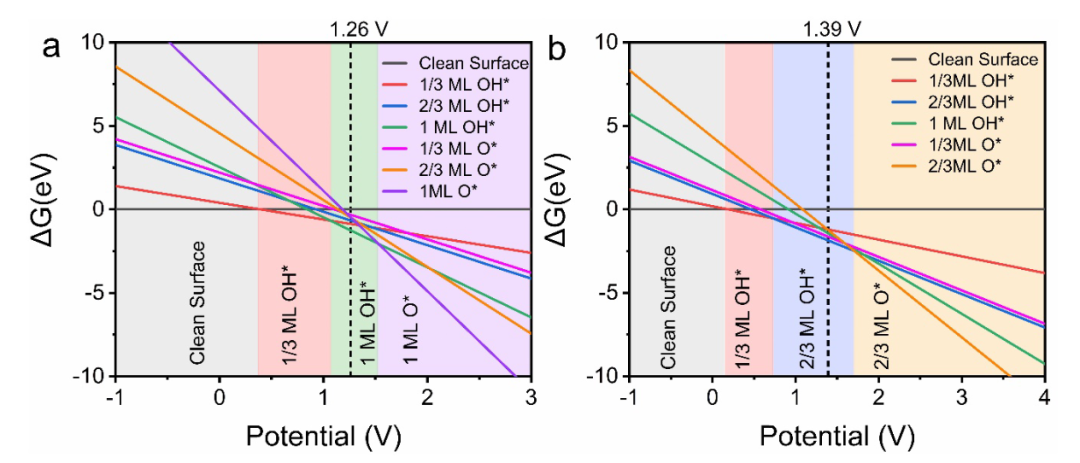
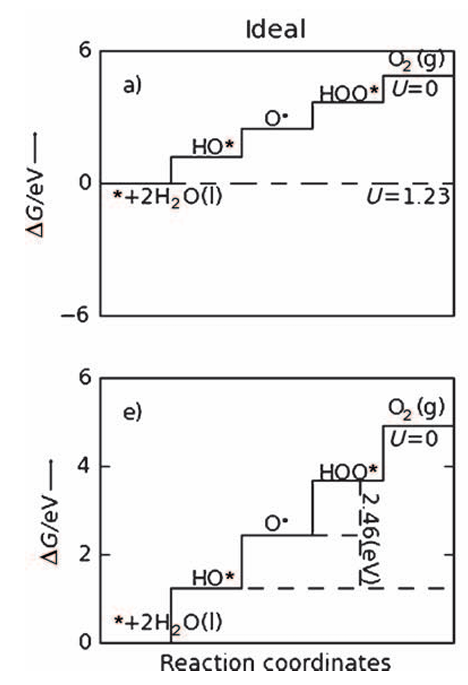
自由能图与过电位的关系是最基础的应用场景,Man等人的研究通过绘制OER自由能图,直观呈现了过电位的起源:理想催化剂的自由能图呈直线,各步骤ΔG均1.23 eV,对应过电位η=0 V,这是理论上的最优状态;而实际催化剂(如IrO₂)的自由能图呈台阶状,*O→OOH步骤的ΔG=2.0 eV,成为速率决定步骤,对应过电位η=0.77 V。
这种关联明确了ΔG_max与过电位的直接对应关系——ΔG_max每增大0.1 eV,过电位约升高0.1 V,为通过优化RDS能垒降低过电位提供了明确目标,例如通过掺杂使O→*OOH步骤的ΔG从2.0 eV降至1.6 eV,过电位可从0.77 V降至0.37 V。
同时,自由能图结合火山图分析,可定位催化剂在活性趋势中的位置,例如RuO₂因ΔG_*OH处于火山图顶点附近,过电位较低,而MnO₂因ΔG_*OH过大,位于火山图左侧,过电位较高。
DOI:10.1002/cctc.201000397
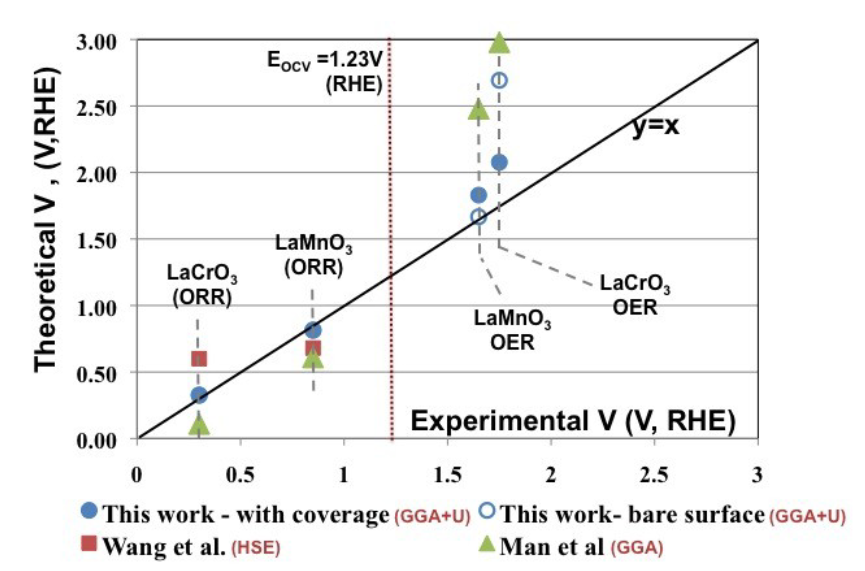
理论与实验过电位的相关性验证是过电位计算可靠性的重要保障,研究团队通过火山图展示了这一关联:数据点包括两种计算条件——蓝色实心点为考虑表面覆盖效应的GGA+U计算结果,蓝色空心点为未考虑覆盖效应的裸表面计算结果。
结果显示,考虑覆盖效应的计算值贴近黑色对角线,误差;而裸表面计算值普遍高估过电位,偏离对角线较远。
这一结果表明,表面中间体的覆盖度会显著影响活性位点的电子态与吸附能——高覆盖度下,中间体之间的排斥作用会弱化吸附强度,使ΔG_max降低,过电位减小,因此理论计算必须纳入覆盖效应校正才能准确预测实验结果。
例如,在Pt (111) 的ORR中,未考虑*OH覆盖度时,理论过电位为0.55 V,而实验值为0.45 V;纳入覆盖度校正后,理论过电位降至0.47 V,与实验高度吻合。
这种理论-实验的一致性验证了过电位计算方法的有效性,同时强调了实际反应条件对理论模型的重要性,为后续计算参数的选择提供了依据。
总结
过电位作为连接催化剂微观吸附能与宏观催化活性的核心描述符,其理论计算的准确性与效率直接决定了电催化理论研究对实验设计的指导价值,而这一目标的实现依赖于精准的吸附能计算、合理的动力学模型与高效的计算工具的协同作用。
未来过电位计算的发展将聚焦于三个方向:动态界面模拟需实现电位依赖的吸附与表面重构的实时耦合,例如通过恒电位AIMD模拟不同电势下催化剂表面的羟基演化,捕捉过电位随结构变化的动态规律。
多尺度耦合需整合电子结构计算、动力学模拟与传质模型,实现从电子转移到宏观电池性能的跨尺度过电位预测,例如在电解槽设计中,同时考虑活性位点过电位与电解液传质过电位,优化整体能量效率;高通量筛选框架的优化需发展更通用的描述符,提升模型对新型催化剂的泛化能力,减少对DFT训练数据的依赖。
这些发展将推动过电位计算从“解释实验”向“预测性能”再到“设计材料”跨越,为开发低过电位、高稳定性的电催化剂提供更强大的理论支撑,加速电催化技术在燃料电池、电解水、CO₂转化等领域的产业化应用。