

WIEN2k是用密度泛函理论计算固体的电子结构的商业收费软件,在Google Scholar上有大约12000个引用。
Wien2K耗时,但是更精确,因此预测电子结构,化学键比较准确,其他性质的计算也很精确。体系不大,可选Wien2k。但相关学习资料比较少,可以说是非常难找!
下载方式:长按识别下方二维码,对话框回复关键词“2k”,免费下载!


Wien2k基于键结构计算准确的方案——势能(线性)增广平面波((L)APW)+局域轨道(lo)方法。在密度泛函中可以使用局域(自旋)密度近似(LDA)或广义梯度近似(GGA)。
第1课:密度泛函理论(DFT)和缀加平面波+局域轨道方法
第2课:写给初学者的WIEN2k软件包综述:初始化、电子自洽迭代、电子密度、态密度以及能带
第3课:结构文件、k点、多种输入文件、AIM
第4课:力、结构优化、超胞、表面和声子
第5课:磁性
第6课:更为先进的处理交换关联势的方法:杂化泛函、LDA+U以及GW
第7课:光学性质、芯能级谱(XAS, EELS)以及BSE
第8课:WIEN2k的安装、并行以及大规模应用
第9课:瓦尼尔函数、宏观极化以及相关性质
第10课:相对论效应和非共线磁性(NCM)
第11课:超精细相互作用以及如何用WIEN2k进行处理

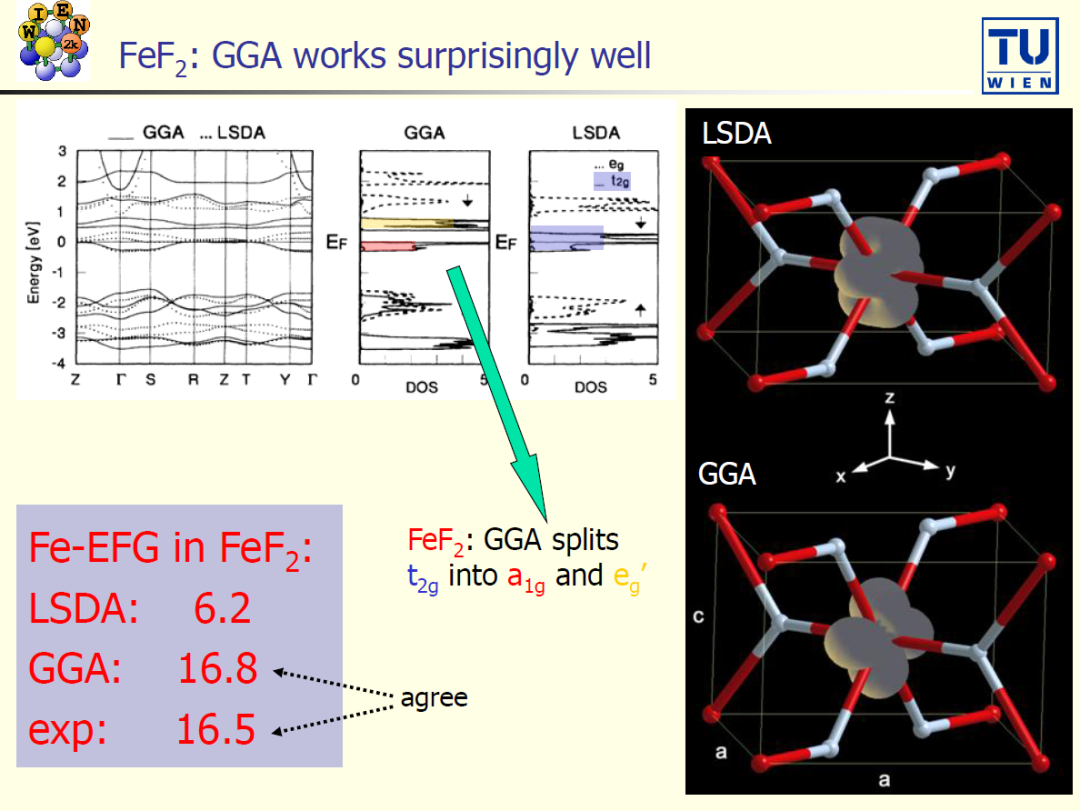

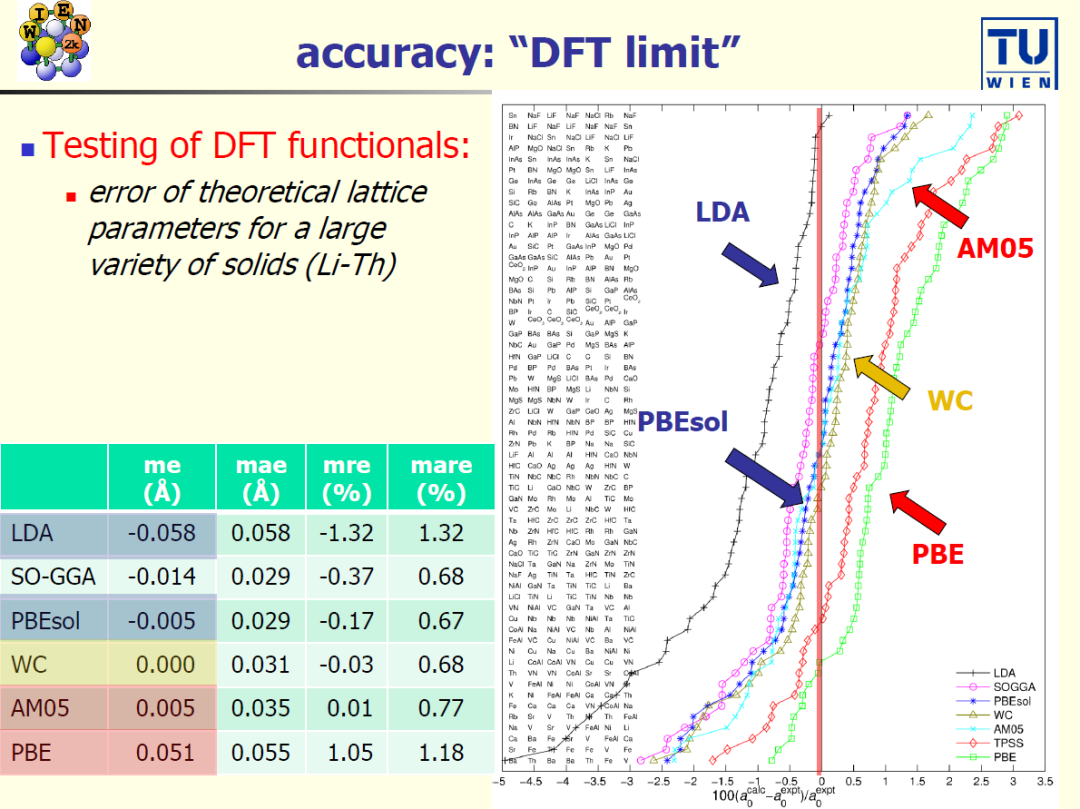


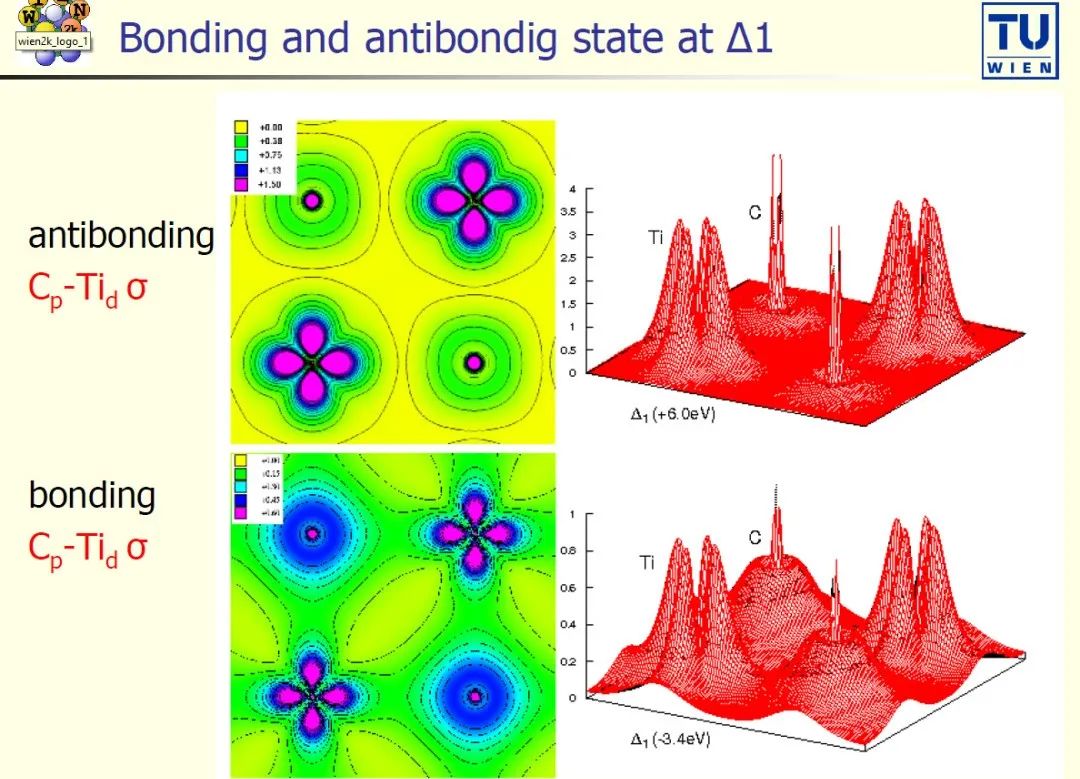
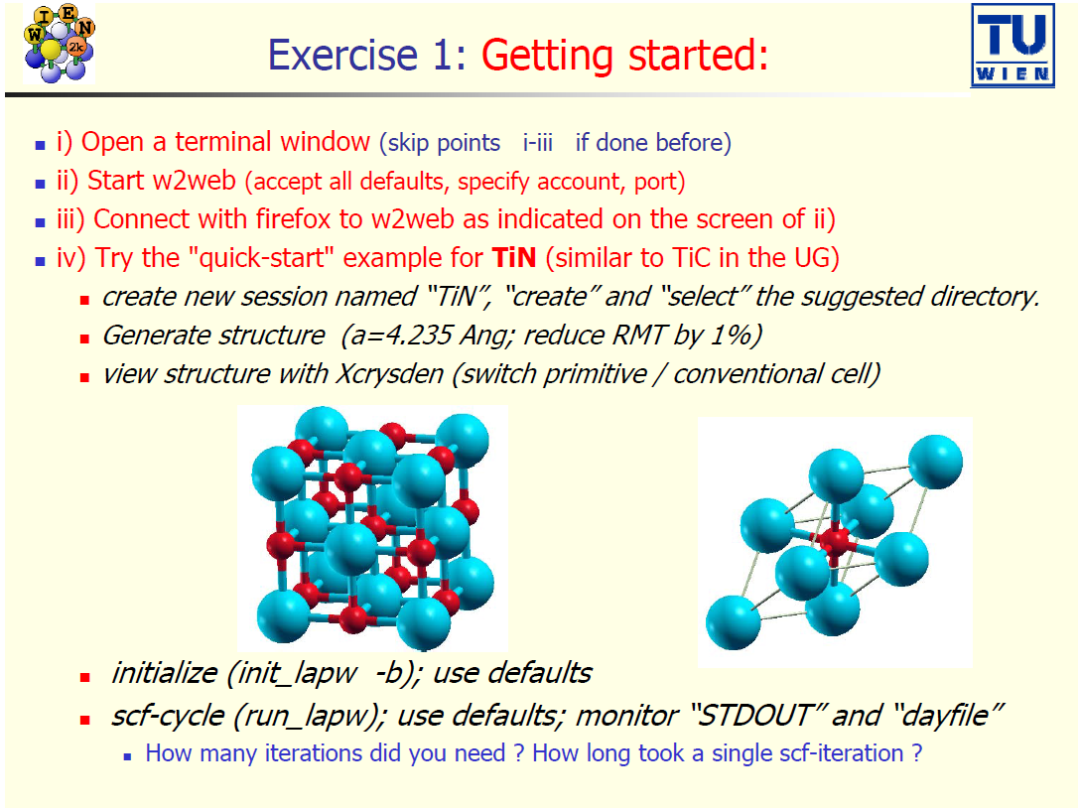
案例 1:开始
案例 2:TiC的晶格常数
案例 3:Mg(OH)2原子位置的优化
案例 4:建立超胞
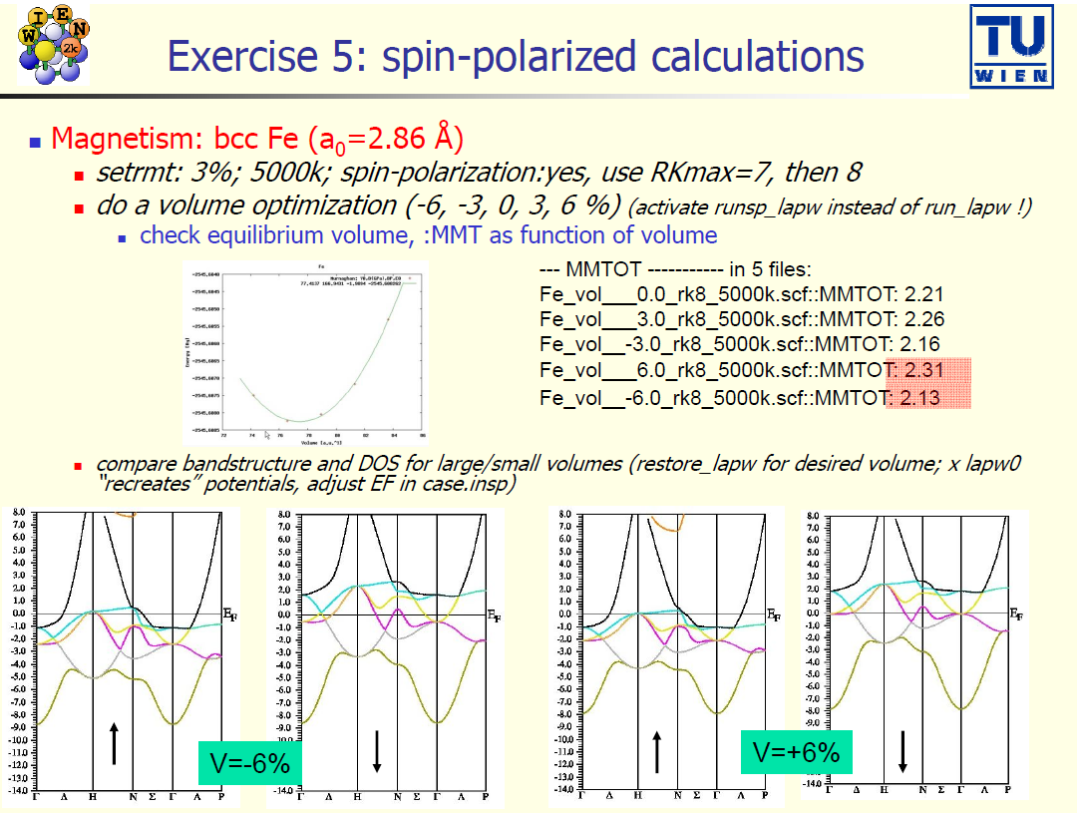
案例 5:自旋极化计算
案例 6:反铁磁计算
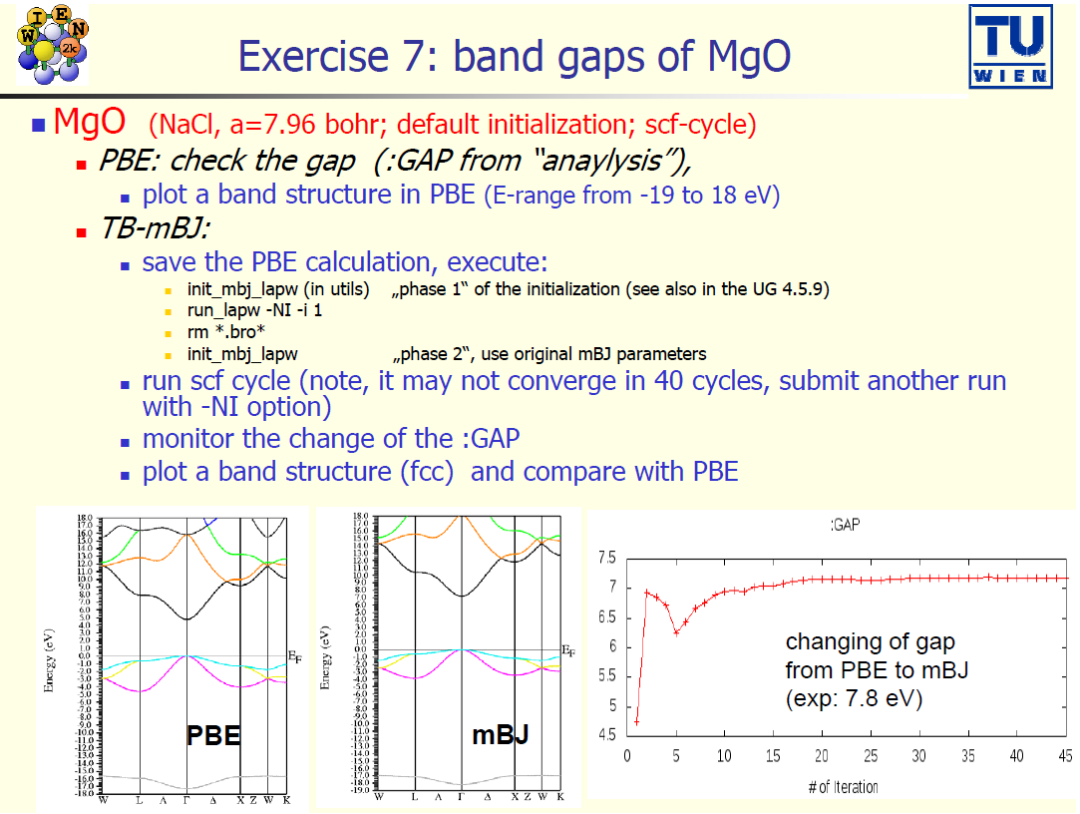
案例 7:MgO的带隙
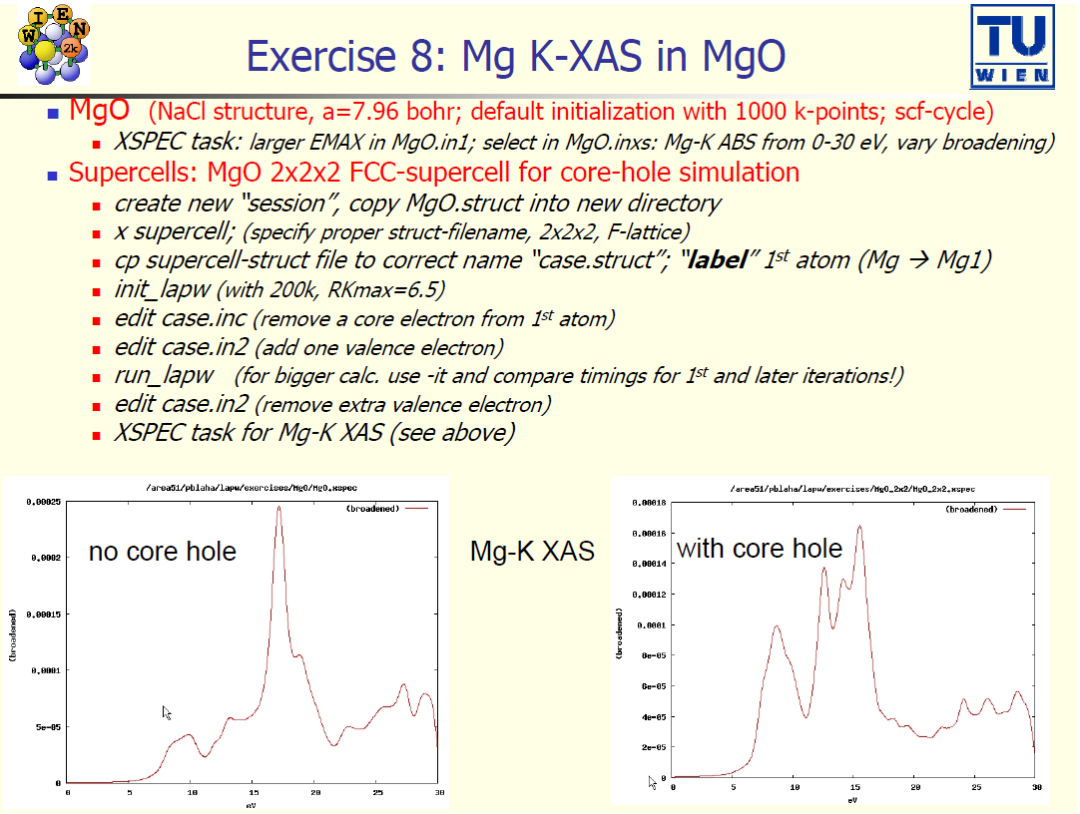
案例 8:MgO中Mg的K-XAS
案例 9:LDA+U计算
案例 10:光学性质
案例 11:SrTiO3的声子








课程所有视频、课件和练习资料,已上传百度网盘。长按识别下方二维码,对话框回复关键词“2k”,免费下载!
先收藏!免费获取全部课程资料
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!