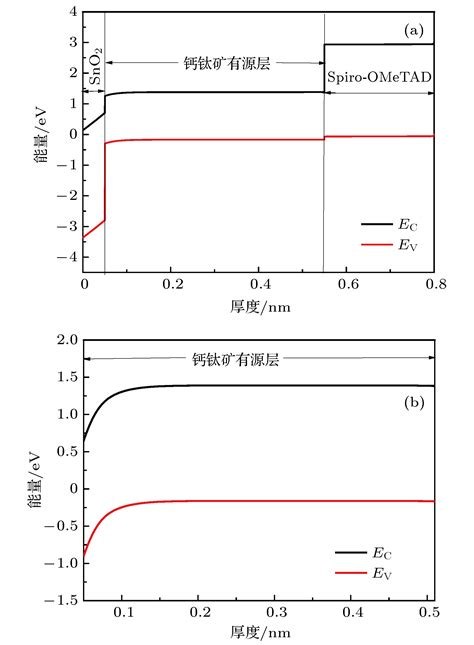
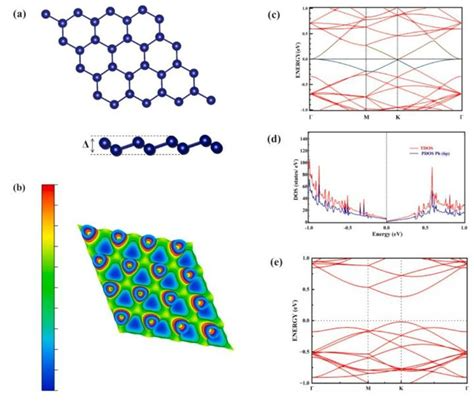
钙钛矿材料的能带结构是其光电性能的核心决定因素之一,其研究对于理解材料的物理特性、优化其应用性能以及开发新型钙钛矿器件具有重要意义。
以下将从钙钛矿的能带结构定义、影响因素、能带工程、以及不同维度钙钛矿的能带特性等方面展开详细讨论。
钙钛矿材料的能带结构是指电子在材料中可能存在的能量分布范围,包括导带(Conduction Band)和价带(Valence Band)之间的能量差,即带隙(Band Gap)。
带隙的大小直接影响材料的光电性能,例如光吸收能力、载流子迁移率等。钙钛矿的能带结构通常通过第一性原理计算(如密度泛函理论)或实验方法(如紫外光电子能谱UPS)来确定。
钙钛矿材料的能带结构受多种因素影响,包括晶格结构、化学组成、掺杂、缺陷等。例如,卤素离子的替代(如Br⁻替代I⁻)会导致能带结构的变化,从而影响材料的光学性质和稳定性。
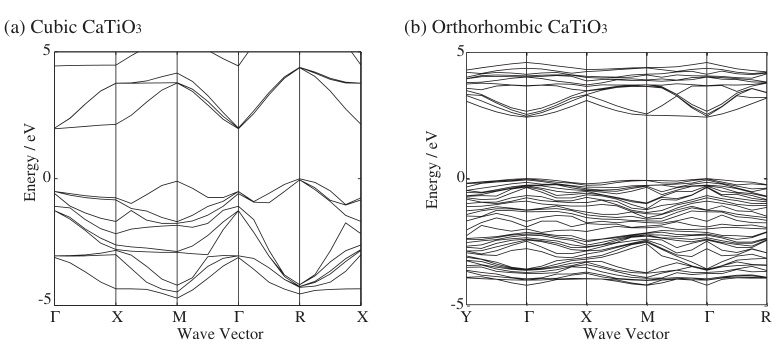
钙钛矿的晶格结构对其能带结构有显著影响。常见的钙钛矿结构包括立方型(CsPbBr₃)和正交型(CsPbBr₃),其中正交结构的能带交叉现象更为复杂,这可能与晶体对称性有关。
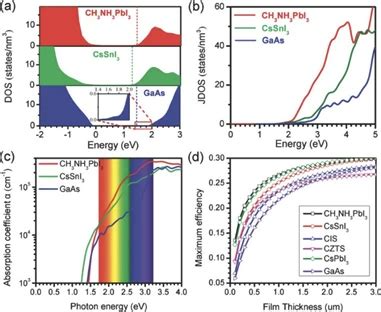
不同的金属离子和卤素离子的引入会显著改变钙钛矿的能带结构。例如,MAPbI₃(甲基铵铅碘化物)的带隙为1.5 eV,而FAPbI₃(氟化铵铅碘化物)的带隙为1.43~1.48 eV,这种差异主要源于不同离子的电子结构和极化效应。
掺杂是调控钙钛矿能带结构的重要手段。通过引入其他金属离子或卤素离子,可以调节钙钛矿的带隙宽度,从而优化其光电性能。例如,锗溴混合掺杂的钙钛矿模型显示其带隙值为1.50 eV,表明掺杂可以实现带隙的精确调控。
缺陷是钙钛矿材料中不可避免的结构不完整性,其对能带结构有重要影响。例如,钙钛矿材料中的铅空位和碘空位会导致能带的分裂和带隙的减小,从而影响材料的稳定性。
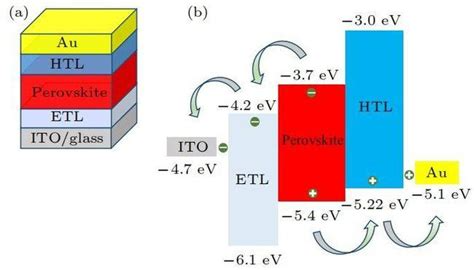
能带工程是通过改变材料的化学组成或结构来调控其能带结构,从而实现特定性能优化的技术。例如,通过引入不同价态的铅离子(如Pb²⁺和Pb⁴⁺)可以调节钙钛矿的带隙宽度,从而实现宽带隙或窄带隙的调控。
钙钛矿的能带结构还可以通过人工量子阱(MQWs)技术进行优化。例如,基于CsPbBr₃的MQWs可以通过引入量子屏障实现I型和II型能带对齐,从而提高器件的光电转换效率。
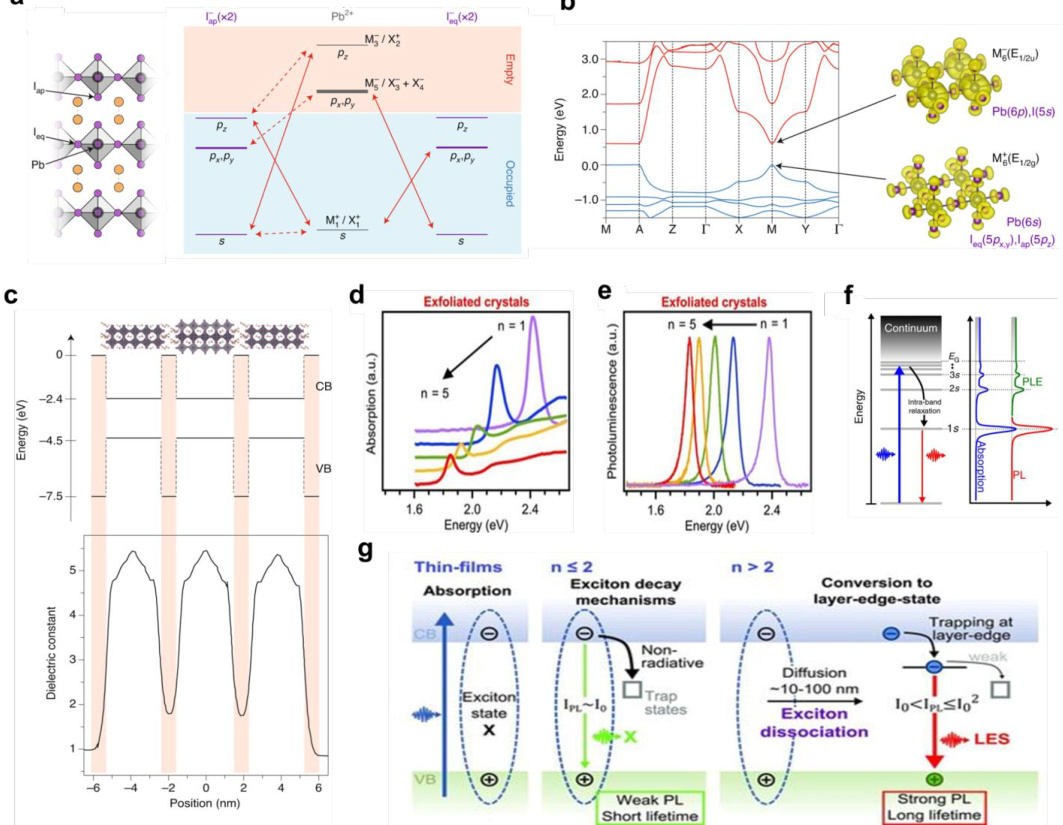
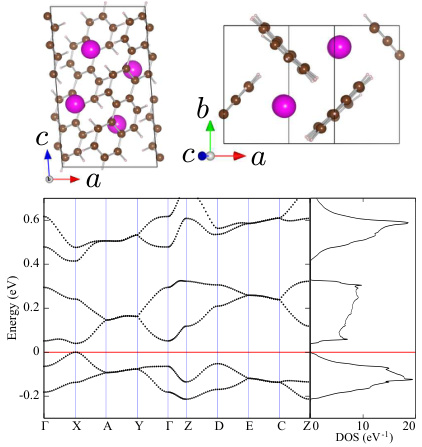
二维钙钛矿(如BA2PbnI3n+1)的能带结构通常表现为较窄的带隙,这与其层状结构和较低的电子散射率有关。研究表明,二维钙钛矿的带隙宽度随着n值的增加而减小,这与其层间耦合强度有关。
三维钙钛矿(如CsPbBr₃)的能带结构较为复杂,其带隙宽度受晶格常数和卤素离子比例的影响。例如,CsPbBr₃的带隙宽度为1.55 eV,而CsPbBr₃的带隙宽度为1.43 eV,表明卤素离子的替代对带隙有显著影响。
低维钙钛矿(如纳米线、量子点)的能带结构通常表现为量子限制效应,导致带隙的增大。这种效应可以用于制备高效率的光电器件。
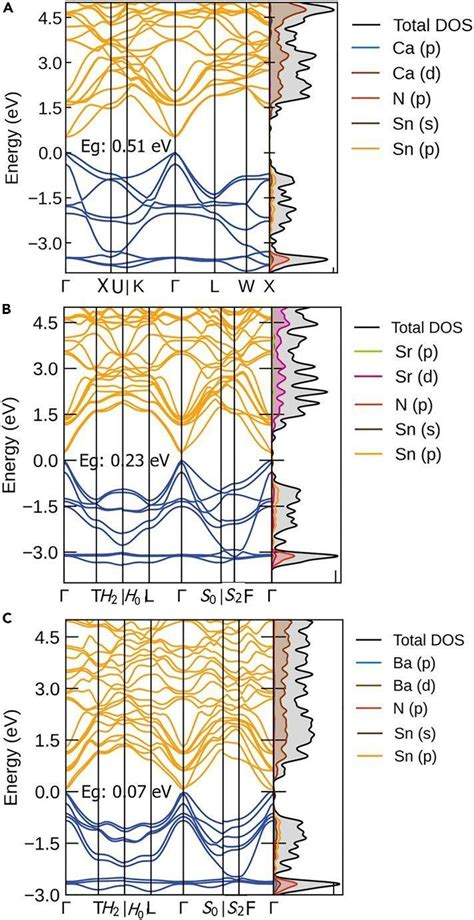
钙钛矿能带结构的实验研究主要通过紫外光电子能谱(UPS)、光致发光光谱(PL)等手段进行。
例如,MAPbI₃的UPS结果显示,其最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)之间的能量差为1.55 eV,这与理论计算结果一致。
理论计算方面,第一性原理计算是研究钙钛矿能带结构的常用方法。通过计算不同k点的能量分布,可以揭示材料的能带交叉和带隙特性。
二维钙钛矿的能带结构计算显示,其带隙宽度在不同k点处存在显著差异,这与其层状结构和电子散射机制有关。
钙钛矿材料的能带结构研究仍处于快速发展阶段。未来的研究方向包括:
通过设计新的钙钛矿结构,如手性钙钛矿和异质结钙钛矿,可以进一步优化其能带结构和光电性能。
通过调控缺陷密度和分布,可以实现对钙钛矿能带结构的精确调控,从而提高材料的稳定性和性能。
通过结合二维和三维钙钛矿的优点,可以开发出具有优异光电性能的复合钙钛矿材料。
钙钛矿材料的能带结构是其光电性能的核心决定因素。通过深入研究其能带结构及其影响因素,可以为钙钛矿材料的优化和应用提供理论基础和技术支持。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!