

电子密度理论通过密度泛函理论(DFT)解析材料的微观电子行为,包括自旋密度(揭示磁性)、电荷密度差(界面电荷转移)、原子电荷(量化电负性)及电子局域化函数(ELF,区分键合类型)。
以COF/MXene异质结为例,曲率调控增强电子离域并优化催化活性。这些工具为材料设计提供原子级电子结构依据,支撑磁性器件、催化反应及储能材料的开发。


电子密度核心概念

密度泛函理论(DFT)以电子密度(ρ(r))为核心变量,通过Hohenberg-Kohn定理将多体问题的基态性质简化为电子密度的泛函,绕过高维波函数计算的复杂性。
Kohn-Sham方程通过构造虚拟的无相互作用电子体系,在等效势场中求解单电子波函数,结合交换–关联泛函(如局部密度近似LDA或广义梯度近似GGA)近似处理多电子相互作用,实现电子密度的自洽计算。
电子密度分析通过衍生量揭示材料的化学与物理特性:差分电荷密度(Δρ)量化界面或反应中的电荷转移(如吸附分子与催化剂表面的电子重分布),自旋密度分布解析磁性材料的局域磁矩(如Fe₃O₄中Fe³⁺的d轨道极化),而电子局域化函数(ELF)则可视化化学键类型(如金属键的离域或共价键的局域特征)。
这类分析工具不仅支撑了从能带结构到催化活性的理论预测,还为材料设计(如调控异质结界面的电荷输运或优化磁性序构)提供了微观电子结构依据,成为连接量子力学计算与宏观性能优化的桥梁。
电子密度本身是三维标量场,但通过不同处理可提取化学键、电荷转移、自旋极化等关键信息。以下为四类核心分析内容:
自旋密度
自旋密度(Δρspin=ρα−ρβ)通过量化自旋向上与向下电子的空间分布差异,解析开壳层体系的磁性与反应活性。
在自旋极化密度泛函理论(DFT)计算中,需采用开壳层基态(如UKS方法)并激活自旋自由度,以捕获未配对电子的分布特征。
正自旋密度区域(Δρspin>0)表征α自旋电子占优,常见于铁磁体(如Fe₃O₄的Fe³⁺位点)或自由基(如苯环π轨道的离域未配对电子,Phys. Chem. Chem. Phys., 2016),而负值区域(Δρspin)对应β自旋主导,揭示反铁磁耦合或单电子转移过程(如O₂在催化剂表面的吸附活化)。
通过Multiwfn等后处理工具可三维可视化自旋密度等值面,并量化原子自旋布居(如Fe³⁺的4.2 μB磁矩),结合过渡态分析揭示磁性材料中自旋极化对反应能垒的调控机制(如自旋相关电荷转移降低CO氧化活化能)。
这类分析为设计自旋电子器件、优化磁性催化剂及解析自由基反应路径提供了关键电子结构依据。
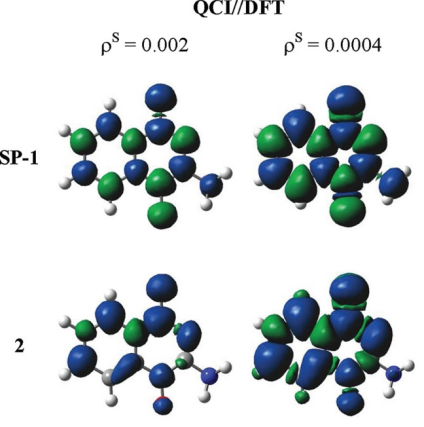
DOI:10.1021/tx2005458
电荷密度差
电荷密度差(Δρ=ρAB−ρA−ρB)通过量化化学键或界面相互作用中的电子重分布,揭示体系的电荷转移机制与成键特性。
Δρ>0区域(如±0.005 e/ų等值面)表征电子聚集,常见于共价键或吸附位点(如CO在催化剂表面的σ键合),而Δρ区域反映电子耗散,对应离子键或电荷转移受体(如Li⁺嵌入电极时的电子损失)。
计算时需优化复合体系AB的几何结构,并对孤立组分A、B进行静态电荷密度计算(统一K点网格与截断能),通过VASP的CHGCAR文件差分处理(如chgdiff.pl脚本)生成Δρ数据,再利用VESTA软件可视化电子转移路径。
以Ti₃C₂ MXene/COF异质结为例(Nano Today, 2020),电荷密度差显示电子从MXene的Ti原子向COF的富电子基团转移(Δρmax=+0.012 e/ų),增强了对多硫化物的锚定与催化转化活性,推动锂硫电池性能提升。
这类分析结合几何优化与电子结构计算,从原子尺度解析界面耦合、催化活性位点及储能机制,为功能材料的设计与性能优化提供了直观的电子分布证据。
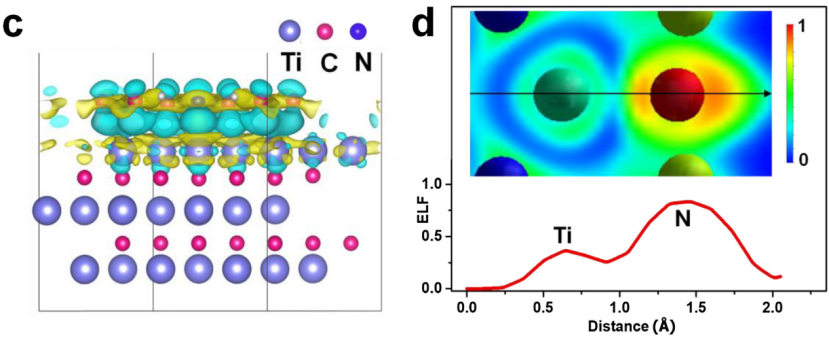
DOI:10.1016/j.nantod.2020.100991
原子电荷
原子电荷作为电子密度在原子尺度上的积分结果,其量化方法各有特色与适用场景。
Mulliken 方法基于原子轨道基函数展开分配电荷,胜在简单快速,却也因对基组敏感而可能产生非物理负电荷,像是一位急性子的 “估算能手”,虽高效却不够严谨。
Hirshfeld 方法通过孤立原子与分子密度的重叠划分电荷,基组依赖性低,却存在低估极化效应的小遗憾,不过改进版 Hirshfeld-I 让精度更上一层楼,宛如在原有方法基础上打了一场漂亮的 “精度升级战”。
ESP 方法则是将分子静电势拟合到原子点电荷模型,适合溶剂化模拟,只是需要大量拟合点且受构象影响,如同在复杂地形中搭建精准的导航系统,虽繁琐却至关重要。
这些方法在实际应用中发挥着重要作用,比如 Hirshfeld 电荷可用于反应活性分析,精准标识亲电 / 亲核位点,成为催化剂活性中心研究的得力助手。
文献中就有实例,2018 年有研究通过 Hirshfeld 电荷量化金属有机框架中金属节点,发现金属离子的电荷极化竟能增强材料对小分子气体的吸附能力,为相关领域研究打开新视角,让我们看到原子电荷量化在揭示物质性质与功能关系中不可或缺的价值。
DOI:10.1039/C8ME00014J
电子局域化函数
电子局域化函数(ELF)是量化电子在空间中局域程度的重要工具,其数学表达式为 ELF (r)=1/[1+[D (r)/D₀(r)]²],其中 D (r) 代表实际电子密度梯度,D₀(r) 为均匀电子气的对应梯度值,二者的比值通过平方反比关系决定了电子的局域化程度 ——ELF 值越趋近于 1,电子在该区域的局域性越强,反之则越趋近于自由电子气的行为。
从可视化特征来看,当 ELF 值高于 0.75 时,往往对应孤对电子或共价键的强局域化区域,这类区域在分子结构中如同 “电子的稳定居所”,承载着原子间成键的关键信息。
例如在典型分子结构中,红色高亮的高 ELF 区域常精准定位共价键或孤对电子的分布;而当 ELF 值接近 0.5 时,体系呈现类自由电子气的行为,这恰是金属键的特征 —— 电子在原子间自由离域,形成维系金属结构的 “电子海洋”。
通过 ELF,研究者不仅能直观观测电子在分子或固体中的分布模式,更能借此区分不同化学键类型(如共价键的局域性与金属键的离域性),为理解化学反应中的电子转移、材料的键合特性乃至催化活性位点的电子结构提供关键线索,成为连接微观电子分布与宏观物质性质的重要桥梁。
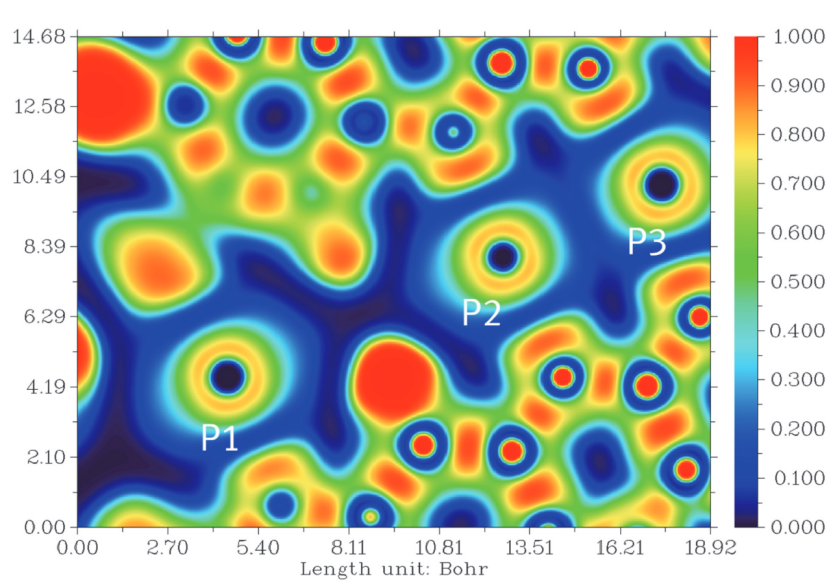
DOI:10.1039/D2DT01762H
经典案例:COF/MXene的电子密度研究
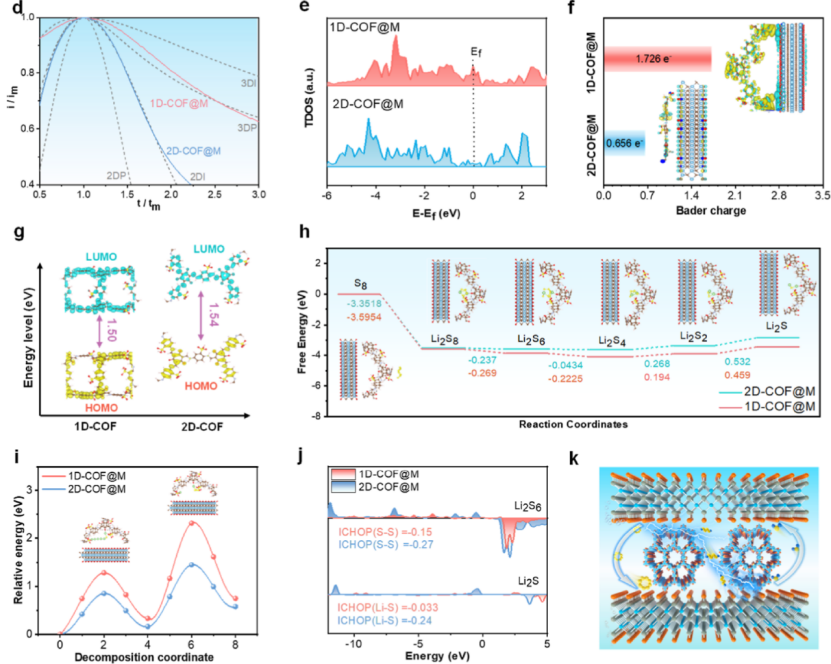
该研究通过密度泛函理论(DFT)计算系统揭示了曲率效应对COF/MXene异质结构催化活性的调控机制,聚焦于电子离域化与硫氧化还原反应动力学的内在关联。
通过构建1D-COF@MXene(高曲率)与2D-COF@MXene(低曲率)模型,计算表明曲率增加显著增强了COF骨架的电子离域化,具体表现为1D-COF@MXene在费米能级附近的态密度(DOS)分布更宽且强度更高,表明其具有更强的电荷传输能力。
Bader电荷分析显示,MXene向1D-COF的电荷转移量(1.726 e⁻)远超2D-COF(0.656 e⁻),表明高曲率结构通过增强界面耦合促进电荷再分布,激活了催化位点的电子活性。
分子轨道分析进一步揭示,1D-COF的LUMO轨道主要集中在C=O基团附近,而HOMO轨道则呈现全局离域特征,其带隙(1.50 eV)较2D-COF(2.10 eV)显著缩小,降低了电荷转移能垒,优化了硫还原反应(SRR)的动力学过程。
吉布斯自由能计算表明,1D-COF@MXene对硫物种转化的各步骤自由能变化(ΔG)均低于2D-COF@MXene,尤其是Li₂S₂→Li₂S的决速步骤能垒从2D体系的0.82 eV降至0.21 eV,归因于曲率诱导的电子离域增强了Li-S键的轨道杂化与断裂能力。
晶体轨道哈密顿布居(ICOHP)分析进一步量化了催化界面与中间体的相互作用:1D-COF@MXene对LiPSs的Li-S和S-S键的ICOHP值较2D体系更弱,表明其通过更强的反键态占据削弱了多硫化物的化学键强度,加速了硫物种的转化与解离。
此外,Li⁺扩散能垒计算显示,1D-COF@MXene沿轴向的扩散势垒(0.32 eV)显著低于2D结构(0.58 eV),证实其分层异质结构通过优化Li⁺传输路径实现了均匀的离子通量分布。
这些理论结果共同表明,曲率调控通过增强电子离域、降低反应能垒、优化轨道相互作用三重机制,协同提升了COF/MXene异质结构对硫氧化还原反应的双功能催化活性,为基于电子结构调控的高效催化剂设计提供了原子尺度的理论支撑。
DOI:10.1021/acsnano.4c17087
总结
电子密度理论计算作为连接量子力学微观世界与材料宏观性质的桥梁,借助自旋密度、电荷密度差、原子电荷、电子局域化分析等核心分析,将抽象的量子力学计算结果转化为化学键特征、磁性行为和反应活性的直观物理图像。
其中,自旋密度分析如同为电子的自旋态绘制“等高线图”,清晰展现材料的磁性分布与自旋极化特性;
电荷密度差通过对比复合体系与孤立组分的电子密度差异,以可视化的色彩梯度(如电子积累的黄色区域与流失的青色区域)精准定位界面电荷转移路径,成为揭示异质结相互作用的“电子显微镜”;
原子电荷量化方法(如 Hirshfeld、Mulliken 电荷)则像给原子 “称量电子重量”,通过积分电子密度定量评估原子的电负性差异,为反应活性位点的识别提供关键参数;
电子局域化函数(ELF)通过量化电子在空间中的聚集程度,直观揭示化学键类型(如共价键、孤对电子)和电子结构特征,是分析材料成键本质的核心理论工具。