

在当今化学催化研究领域,金属有机框架(MOF)材料凭借其独特的结构和优异的性能,成为了研究的热点。针对 MOF 催化计算解决方案的需求,我们将从电子结构分析、性能对比和个性化方法三个层面进行详细阐述,并结合理论建模与计算工具的应用,深入探讨 MOF 催化的内在机制。
电子结构分析
能带与态密度(DOS)
MOF 的电子能带结构和态密度是理解其催化活性的核心要素。能带结构反映了电子在材料中的能量分布情况,而态密度则展示了在不同能量状态下电子的分布密度,它们共同为研究 MOF 的电子行为提供了关键信息。
以 Fe – MOF 为例,其配体(如 NH、O、S)在催化过程中扮演着重要角色。这些配体能够通过电荷再分布的方式,显著地调节 MOF 的带隙和电子结构。在 Fe – O MOF 体系中,研究发现其带隙较窄。较窄的带隙意味着在光照条件下,更容易产生光生载流子,并且有利于光生载流子的分离,从而为催化反应提供更多的活性物种,提升催化活性。
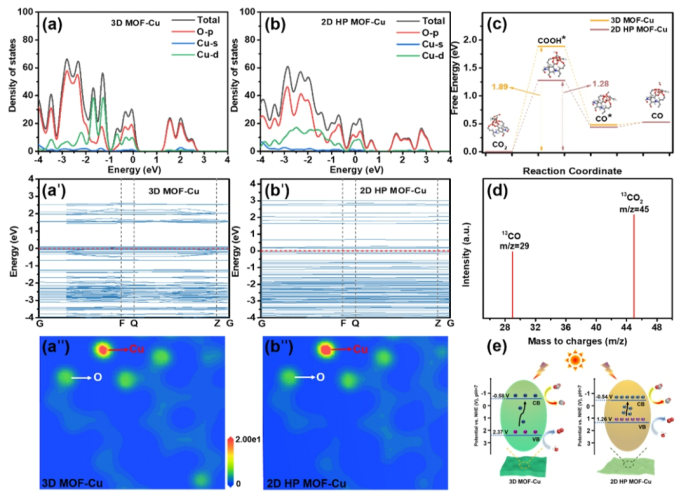

https://doi.org/10.1016/j.apcatb.2024.124567
DFT理论计算表明Cu-MOF 的电荷密度主要分布在铜原子上。引入缺陷后,2D HP MOF-Cu 中更多的电子集中在铜原子附近,使其具有更强的供电子能力,从而促进了CO2的吸附和活化。
2D HP MOF-Cu中配位-OH的引入可以缩小带隙,提高活性位点(Cu)上的载流子密度,降低CO2还原过程中决定速率步骤的自由能垒,同时,2D HP MOF-Cu超薄片层大量暴露的不饱和配位金属活性位点协同提高了CO2的光催化还原效率。
差分电荷密度与 Bader 电荷
Bader 电荷分析是一种量化活性位点电荷转移的有效手段。在多相催化体系中,电荷转移对于理解催化剂的活性和选择性至关重要。
例如,当 Cu 掺杂 CeO₂时,通过 Bader 电荷分析发现,在富 Cu 区域,表层氧空位的形成能降低。氧空位的存在增加了催化剂对 SO₂的吸附能力,因为 SO₂分子能够与氧空位发生相互作用,从而促进了催化反应的进行。
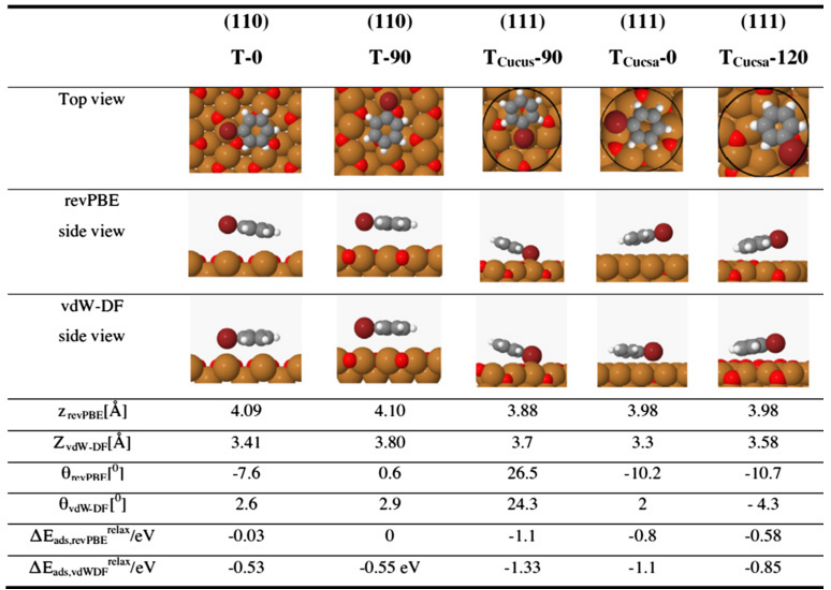
同样地,在 Mo/Co 催化剂体系中,N 掺杂导致 Mo 位点出现电荷局域化现象。这种电荷分布的改变降低了 H₂的吸附能垒,使得 H₂更容易在催化剂表面吸附和活化,为后续的催化反应创造了有利条件。
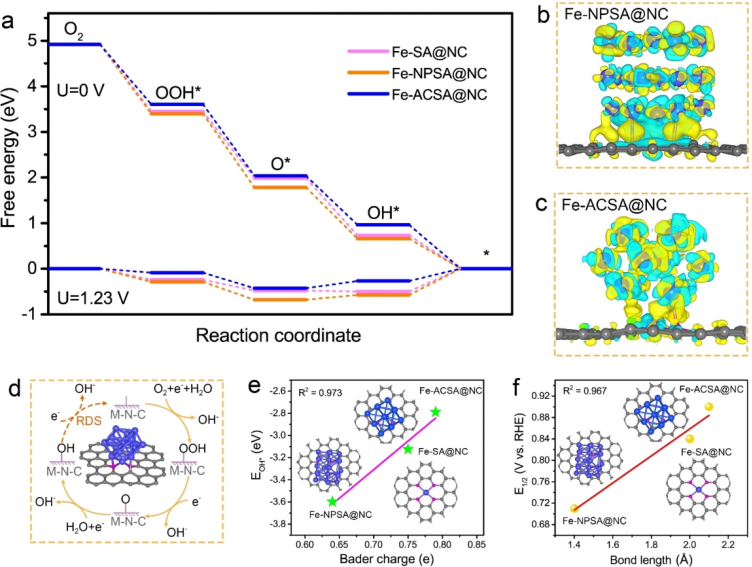
https://onlinelibrary.wiley.com/doi/10.1002/anie.202116068
通过DFT计算来深入研究Fe原子团簇与Fe-N-C之间的协同机制。从图a的ORR自由能图可以看出,在0 V下,所有样品的ORR步骤都是自发的放热反应。而在1.23 V下,可以看出,OH*从活性位点脱附是反应的速率决定步骤(RDS)。
对于Fe-ACSA@NC,RDS需吸热0.2679 eV,并且在三个样品中最低,表明OH*更容易从Fe-ACSA@NC活性位点上脱附。为了进一步揭示电荷再分布的规律,作者进行了差分电荷密度分析,结果如图b-c所示,表明Fe团簇的修饰能够诱导Fe-N活性位点周围发生电子再分布:金属Fe中心周围的电子转移到邻近的碳上,使Fe中心的正电荷增加,削弱了对OH*的吸附。
此外,OH*的吸附能也与Bader电荷呈线性关系,其中,Fe-NPSA@NC和Fe-SA@NC具有较低的Bader电荷(分别为0.64和0.75),OH*吸附能也较低(EOH*分别为-3.60和-3.13 eV),因而表现出较强的OH*结合能。
相反,Fe Fe-ACSA@NC具有最高的Bader电荷(0.79),表现出最弱的OH*吸附能(-2.79 eV),有利于OH*的脱附。此外,Fe-N键长也与ORR半波电位呈线性相关,如图f所示,这与之前的研究报道结果一致。
ELF(电子局域函数)
功函数反映了材料表面电子逸出的能力,它在催化过程中影响着催化界面的电荷转移。而 ELF 分析则专注于描述化学键的共价性或离子性,为研究催化剂的电子结构提供了另一个重要视角。
当 MOF 节点进行功能化处理,如 Ir、Rh 单原子锚定在 MOF 节点上时,通过 ELF 分析可以观察到金属 – 配体键的共价性增强。这种共价性的增强优化了反应中间体在催化剂表面的吸附行为,使得反应中间体能够更稳定地吸附在催化剂表面,从而促进催化反应的进行。
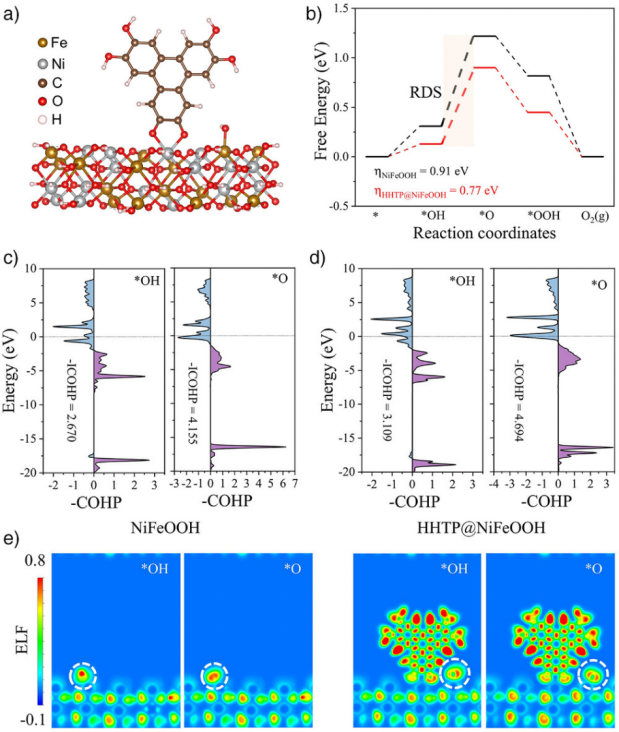
https://doi.org/10.1002/anie.202504148
DFT计算表明:HHTP配体使活性位点从Ni转向Fe(图a),*OH→*O的吉布斯自由能垒从0.91 eV降至0.77 eV(图b)。晶体轨道哈密顿布居(COHP)分析显示,配体增强Fe-O键合态(图c-d),电子局域函数(ELF)证明中间体与Fe位点的电子离域作用(图e)。
功函数
功函数作为材料的一个重要物理性质,在 MOF 催化中具有不可忽视的作用。它决定了电子从材料表面逸出的难易程度,进而影响着催化反应中电子的转移过程。
在 MOF 催化体系中,功函数的大小与催化剂的活性和选择性密切相关。例如,当 MOF 与反应物分子相互作用时,功函数的差异会导致电子在界面处的转移,从而影响反应物分子的吸附和活化过程。
如果 MOF 的功函数与反应物分子的电子亲和能匹配得当,就能够促进电子从 MOF 转移到反应物分子上,使得反应物分子更容易被活化,进而提高催化反应的效率。
因此,精确测量和调控 MOF 的功函数,对于优化 MOF 催化剂的性能具有重要意义。在实际研究中,可以通过改变 MOF 的组成、结构或者表面修饰等方法来调节其功函数,以满足不同催化反应的需求。

https://doi.org/10.1039/D4QI03353A
采用“离子交换–水解–热解”策略制备了 Ru-RuO₂-ZnO 异质结构。在碱性 HER 中,随中空菱十二面体 Ru-ZIF-8 前驱体的热解温度从 880℃升高至 920℃,其催化活性先升高后降低。最佳的 -Ru-ZnO-900 催化剂在 10 mA cm⁻² 电流密度下过电位仅为 25.0 mV,且在 100 mV 过电位下的 H₂转换频率 (TOF) 达 0.23 s⁻¹。
-Ru-ZnO-900 催化剂在 10 mA cm⁻² 电流密度下过电位仅为 25.0 mV,且在 100 mV 过电位下的 H₂转换频率 (TOF) 达 0.23 s⁻¹。
这种优异的性能源于具有适宜氧空位 (Ov) 含量的 Ru⁰{12̅0}-RuO₂{2̅21}-ZnO{001}异质界面,其通过以下协同效应发挥作用:1. 降低功函数且使带中心下移;2. 加快 H₂O 吸附–解离动力学;3. 适度减弱催化剂表面对 H*的过强电子相互作用;4. 优化 H*吸附与 H₂脱附间的平衡;5. 提高决速步 (H₂脱附) 速率并降低 HER 表观活化能。
研究发现,过电位与 H*吸附吉布斯自由能变化 (ΔGH*) 呈线性关系,揭示 轨道调控对催化活性的关键作用。该工作为构建具有可控功函数和带中心的异质界面提供了新策略,对清洁能源转换催化剂的设计具有重要意义。
轨道调控对催化活性的关键作用。该工作为构建具有可控功函数和带中心的异质界面提供了新策略,对清洁能源转换催化剂的设计具有重要意义。
性能对比参数
吸附能与台阶图
吸附能是评估 MOF 催化活性的关键指标之一。它反映了反应物分子与催化剂表面之间相互作用的强弱程度。以 Ni₂V – MOFs 为例,通过 V 掺杂能够调控 Ni 位点的 d 带中心。这种调控使得 OER(氧析出反应)决速步骤(*OH → *O)的吸附能降低,从而显著提升了催化剂的性能。实验数据表明,该催化剂的过电位仅为 244 mV,展现出了优异的催化活性。
台阶图,也称为自由能图,它能够直观地展示多步反应的能垒分布情况。通过观察台阶图,研究人员可以清晰地了解到整个催化反应过程中每一步的能量变化,从而确定决速步骤,为优化催化剂提供重要依据。例如,在 Fe – NH MOF 催化的 ORR(氧还原反应)中,通过台阶图分析发现其过电位为 0.38 V,优于 Fe – O 和 Fe – S 体系,这表明 Fe – NH MOF 在 ORR 反应中具有更好的催化性能。
d 带中心理论
d 带中心理论在解释过渡金属催化活性方面具有重要意义。d 带中心位置决定了过渡金属对反应物的吸附强度。
在 Co 功能化 MOF 体系中,Co 的 d 带中心上移。这种变化增强了 MOF 对丙烷 C – H 键的活化能力,使得丙烷分子更容易在催化剂表面发生反应,从而提升了氧化脱氢反应的选择性。
类似地,在 Ni 掺杂 Bi₂MoO₆体系中,通过 d 带中心调控能够优化材料的光吸收和载流子分离效率。这是因为 d 带中心的改变影响了材料的电子结构,使得光生载流子更容易产生和分离,进而提高了光催化反应的效率。
https://pubs.acs.org/doi/10.1021/acsnano.3c02749
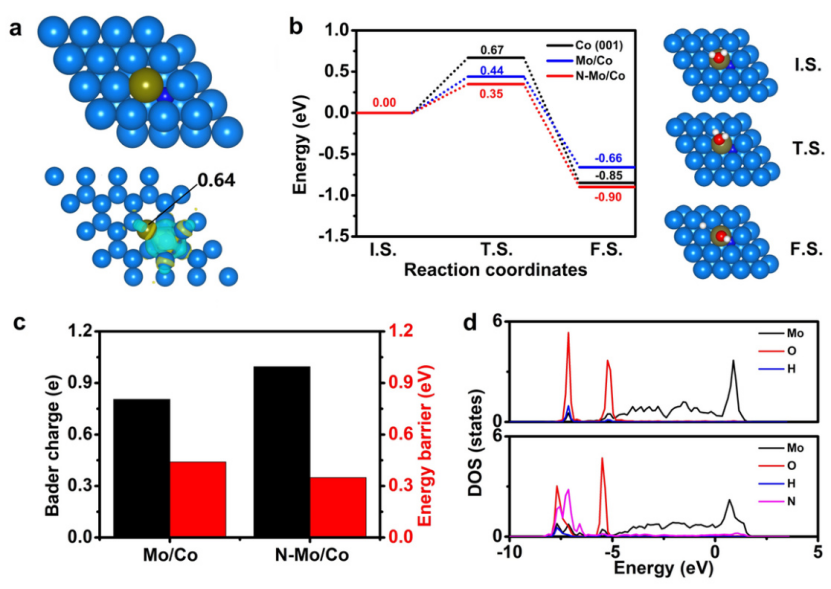
为了了解Rh节点对于Ni-BDC MOF晶体结构和电子结构的影响,作者首先利用DFT求其进行了理论计算。结果表明,Rh节点的引入使两个相邻的Ni-O键长缩短,这将有利于提升催化剂的稳定性。
另外,当原子Rh被引入Ni-BDC时,Rh节点附近发生了强烈的电荷密度重新分布,使Ni-BDC电导率增加。NiRh-BDC的d带中心(Ed)为-1.48 eV,高于Ni-BDC(-2.47 eV)。根据d带理论,当催化剂的Ed上移时,EF以上氢的反键态将增加,从而增强催化剂对中间体的亲和力,这对于提高催化剂性能至关重要。
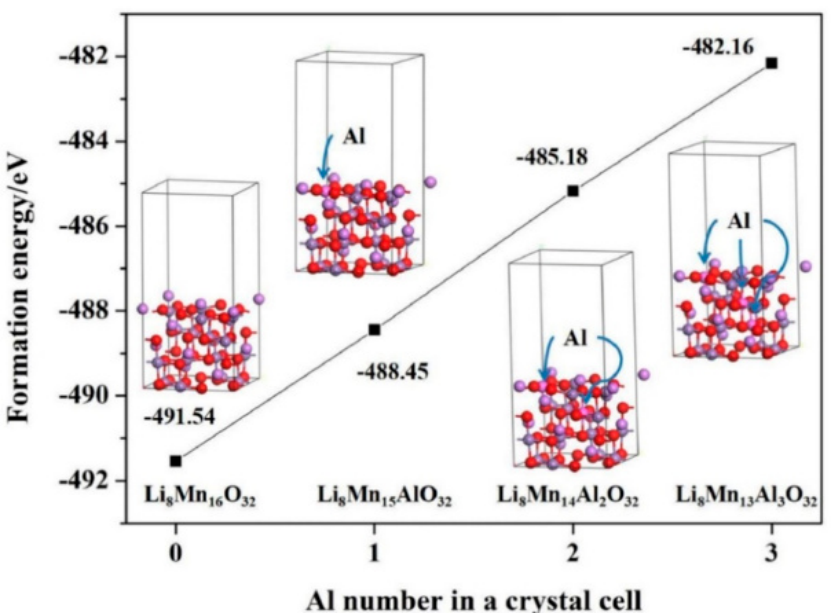
掺杂能与形成能
掺杂能和形成能的计算在指导 MOF 材料设计方面发挥着关键作用。
当 V 引入 CoFe – LDH 中时,计算得到的形成能较低,这意味着 V 的掺杂能够使体系更加稳定。同时,V 的掺杂优化了 Co²⁺/Fe³⁺的电子结构,降低了 OER 过电位,提高了催化剂的性能。
在 Hf – MOF 体系中,Hf – O 键的形成能较低。这种低形成能促进了异丙醇羟基 H 的转移,使得反应势垒仅为 45.4 kcal/mol,有利于催化反应的进行。通过对掺杂能和形成能的计算,研究人员可以预测不同元素掺杂对 MOF 性能的影响,从而有针对性地设计和合成具有优异性能的 MOF 催化剂。
个性化分析方法
过渡态搜索(TS)与势垒计算
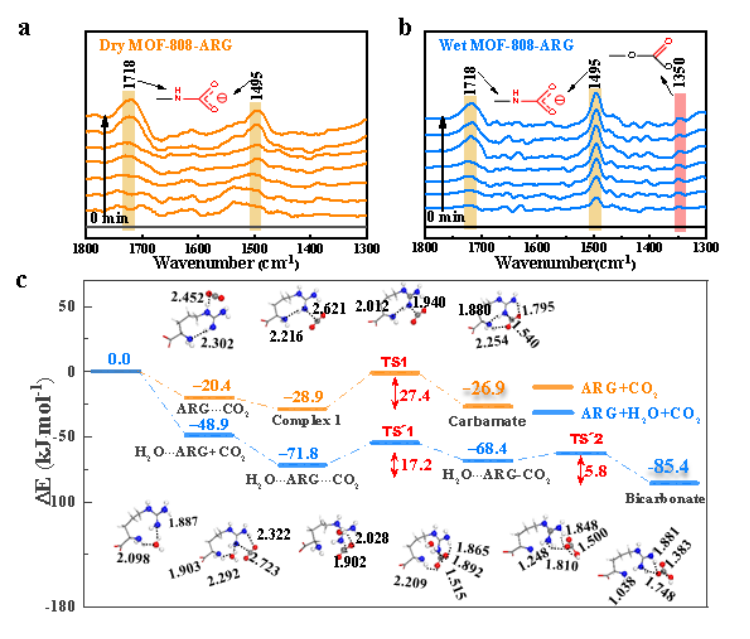
在研究 MOF 催化反应机理时,确定反应路径和计算反应势垒是至关重要的。使用 CI – NEB( climbing image nudged elastic band method)或微动弹性带法(如 VASP 内置)可以有效地确定反应路径。以 Hf – MOF 催化环己酮还原反应为例,通过多坐标驱动法能够准确找到过渡态,即反应过程中的最大能量点。
该反应的势垒为 45.4 kcal/mol,这一数据为评估反应的难易程度提供了重要依据。同样地,在 CO₂在 COP – S 上的还原反应中,通过过渡态搜索发现 * COOH 形成是决速步,并且该步骤的能垒最低。这一结果对于理解 CO₂还原反应的机理和优化催化剂具有重要意义。
https://doi.org/10.1007/s12274-022-5028-5
COOP/COHP 分析
晶体轨道重叠布居(COOP)和晶体轨道哈密顿布居(COHP)是分析化学键强度的有力工具。
在 Fe – O MOF 体系中,通过对 Fe – O 键的 COHP 分析发现,该键存在强反键态。强反键态的存在削弱了 Fe – O 键的强度,使得 O 活性物种更容易生成,从而促进了催化反应的进行。通过 COOP/COHP 分析,研究人员可以深入了解 MOF 中化学键的性质,为解释催化活性和选择性提供理论支持。
https://doi.org/10.1002/anie.202504148
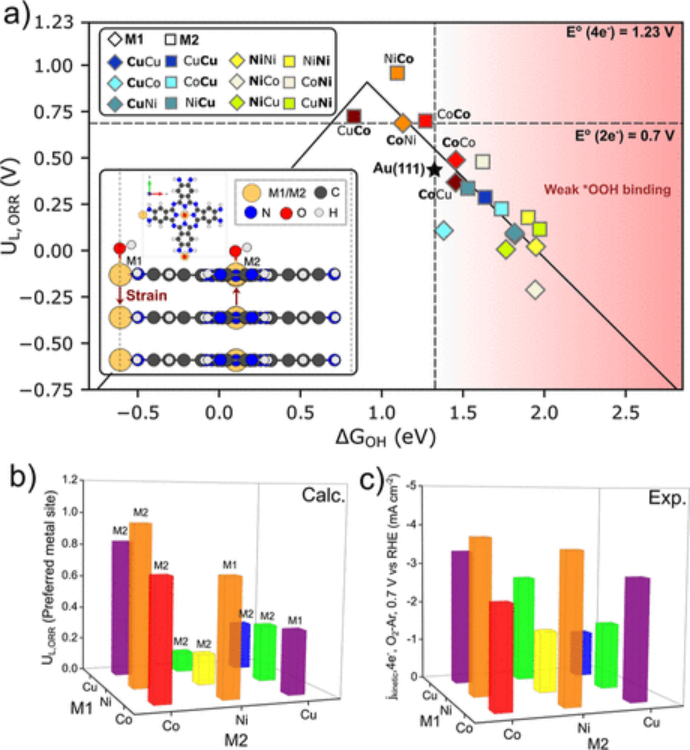
火山图
火山图通过关联描述符(如ΔG_H*)与活性趋势,为研究催化剂的性能提供了直观的可视化工具。
以 Pt (111) 的氧还原反应为例,其氧还原过电位源于 O/OH 中间体的强吸附。通过火山图分析发现,当 ΔG_H* ≈ 0 时,对应着最优的催化活性,即火山图的顶点位置。这一结果为筛选和设计高效的氧还原催化剂提供了重要的理论指导。
https://doi.org/10.1021/jacs.4c02229
AIMD 模拟
AIMD 模拟作为一种先进的计算方法,在 MOF 催化研究中具有独特的优势。它基于第一性原理,能够在考虑电子与原子核相互作用的基础上,模拟分子体系在一定温度和压力条件下的动态行为。
在研究 MOF 催化反应时,AIMD 模拟可以提供许多重要信息。比如,在模拟 MOF 与反应物分子相互作用的过程中,可以观察到反应物分子在 MOF 表面的吸附过程,包括吸附位点的选择、吸附构型的变化以及吸附能的动态变化等。同时,AIMD 模拟还可以研究反应过程中分子的振动、转动等微观运动,这些信息对于理解催化反应的动力学过程至关重要。
此外,通过 AIMD 模拟还可以研究温度、压力等外界条件对催化反应的影响,为优化反应条件提供理论依据。在实际应用中,AIMD 模拟与实验相结合,可以更深入地揭示 MOF 催化反应的本质,推动 MOF 催化领域的发展。
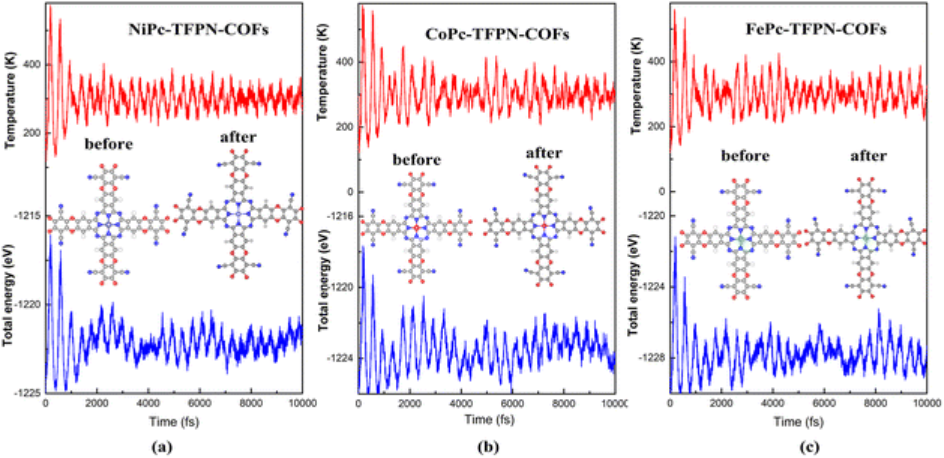
如图所示,通过VASP软件进行了AIMD模拟,探究MPc-TFPN-COFs的能量和温度随模拟时间的变化,发现其结构只有略微变化,表明MPc-TFPN-COFs光催化剂具有良好的稳定性,有利于实验制备。
工具与泛函选择
DFT 方法
在 MOF 催化计算中,密度泛函理论(DFT)是常用的计算方法。在计算吸附能和进行结构优化时,优先选用 GGA – PBE 或 PW91 泛函。这两种泛函在描述电子 – 电子相互作用方面具有较好的性能,能够准确地计算出吸附能和优化 MOF 的结构。
而在精确计算带隙时,HSE06 或杂化泛函则更为适用。这些泛函能够更准确地描述电子的交换 – 关联能,从而得到更精确的带隙值,为研究 MOF 的电子结构提供可靠的数据。
软件平台
目前,有多种软件平台可用于 MOF 催化计算。VASP 适用于周期性体系的计算,它能够高效地处理具有周期性结构的 MOF 材料,在结构优化、电子结构分析等方面具有强大的功能。
Gaussian 则常用于簇模型的计算,它可以对 MOF 中的局部结构进行精确的量子化学计算,为研究 MOF 的微观结构和电子性质提供详细信息。
CP2K 在 AIMD 模拟方面表现出色,它能够在原子尺度上模拟分子的动态行为,为研究 MOF 催化反应的动态过程提供有力支持。
后处理工具
为了更好地分析计算结果,需要使用一些后处理工具。VASPKIT 可以进行能带和态密度分析,帮助研究人员直观地了解 MOF 的电子结构特征。
Bader 电荷工具用于计算 Bader 电荷,量化活性位点的电荷转移情况。
VESTA 则是一款强大的可视化工具,它能够将计算得到的结构和电子密度等数据以直观的图形方式展示出来,方便研究人员观察和分析。
总结
MOF 催化计算是一个复杂而又充满挑战的领域,需要综合运用电子结构分析(如能带、DOS、Bader 电荷分析)、性能参数(如吸附能、d 带中心)与高级方法(如过渡态搜索、COHP 分析、AIMD 模拟)。通过多尺度建模,能够深入揭示活性位点的作用机制,为设计和开发高效的 MOF 催化剂提供理论指导。
同时,实验与计算协同发展是未来的关键方向。例如,火山图可以指导实验中的材料筛选,而实验结果又可以验证计算模型的准确性,两者相互促进,共同推动 MOF 催化领域的发展,为实现更高效、更绿色的催化反应奠定基础。