分子动力学模拟(MD)和密度泛函理论(DFT)是计算化学领域常用的两种重要工具,广泛应用于物质的结构、性质以及反应机制的研究。
尽管它们在模拟对象上有很多交集,但它们在原理和应用上有着明显的区别。本文将从原理和应用两个方面对MD和DFT进行对比,并通过具体的计算案例来说明它们的应用场景。
原理对比
分子动力学模拟(MD)
分子动力学模拟是一种基于经典力学的计算方法,主要通过求解分子间的相互作用力来模拟粒子(分子、原子)随时间演化的过程。MD的核心原理是通过牛顿运动定律计算出粒子的加速度,从而得到它们的运动轨迹。模拟过程中涉及到的主要参数包括原子间的势能函数(通常是经典力场)、粒子的初始速度和位置等。
MD模拟的基本步骤包括:
1、确定分子的初始配置(例如原子位置和速度)
2、计算粒子间的力
3、更新粒子的位置和速度
4、重复计算,得到粒子的运动轨迹

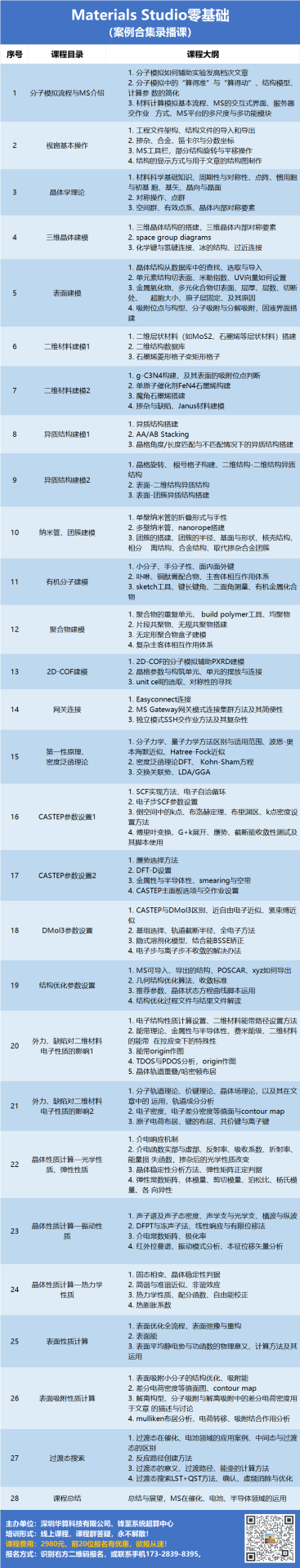
分子动力学模拟轨迹
密度泛函理论(DFT)
密度泛函理论主要通过求解电子密度来描述分子系统的电子结构。DFT的核心原理是通过变分原理对电子密度进行优化,求解哈密顿量(包含动能、势能等)来获得系统的基态能量和其他物理量。DFT不直接计算波函数,而是通过电子密度的泛函来描述体系的总能量,从而显著简化了计算复杂性。
DFT计算的步骤通常包括:
1、假设一个初始电子密度
2、使用Kohn-Sham方程计算电子结构
3、优化电子密度直到达到收敛
4、计算体系的能量、结构及其他性质
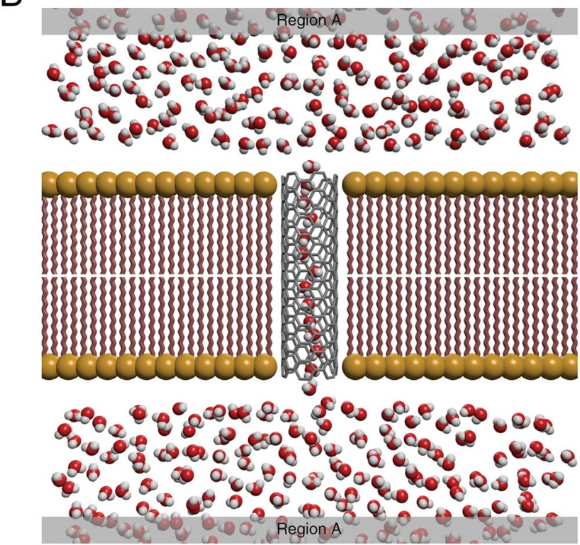
DFT计算的电荷密度
应用对比
分子动力学模拟(MD)的应用
MD模拟广泛应用于研究分子在不同条件下的动态行为,适用于大规模、长时间尺度的模拟。其常见应用领域包括:
材料科学:研究材料的结构演化、力学性能(如断裂韧性、热导率等)
生物分子模拟:研究蛋白质、DNA、RNA等生物大分子的折叠、相互作用、反应动力学
化学反应动力学:通过模拟分子碰撞和反应路径,研究反应机制
液体和气体模拟:研究液体、气体的物理性质如粘度、扩散系数等
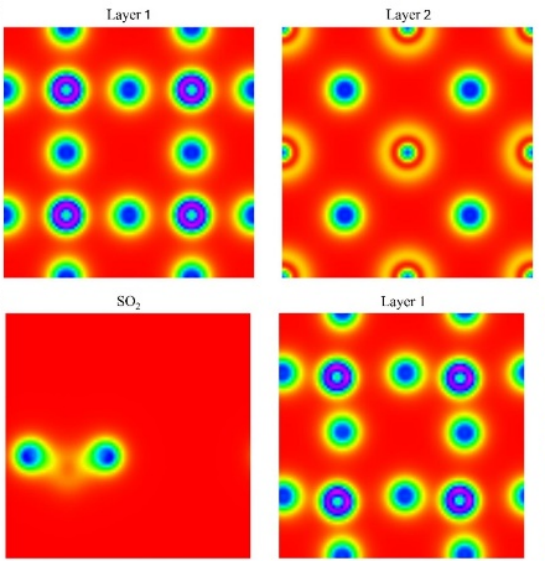
密度泛函理论(DFT)的应用
DFT主要用于研究分子和固体系统的电子结构及其相关性质,特别是基态能量和电子密度的计算。它通常用于研究:
分子电子结构:通过DFT计算分子的基态能量、分子轨道、反应路径等
固体材料的性质:计算固体的带隙、晶格常数、压电效应等
催化反应研究:研究催化剂表面与反应物的相互作用、反应能垒
光电材料设计:计算材料的光吸收、光致发光等性质
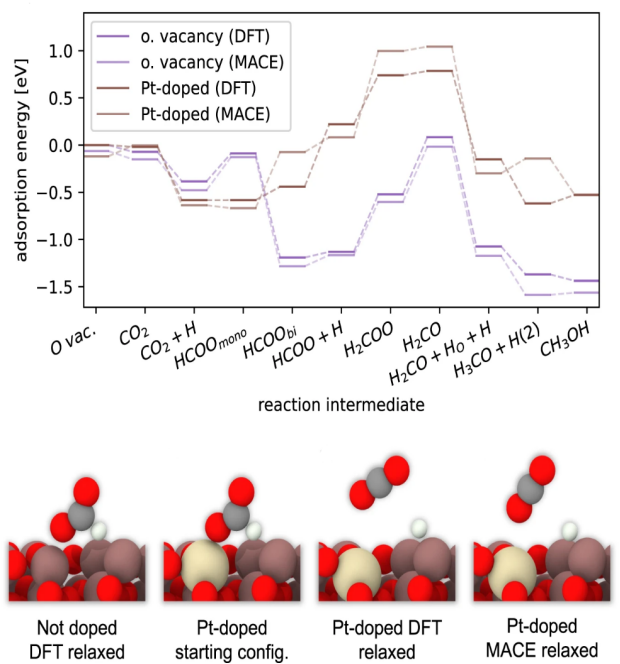
MD和DFT计算案例
MD计算案例:模拟水分子在不同温度下的扩散行为
在材料科学领域,MD模拟可以用来研究液体分子在不同温度下的扩散行为。例如,通过MD模拟水分子在不同温度下的运动轨迹,可以计算水的扩散系数,并进一步分析温度对水分子运动的影响。
步骤:
选择水分子模型和力场(如TIP3P力场)
设置初始温度和压力,使用常见的温度控制算法(如Nosé-Hoover thermostat)
运行模拟,收集水分子的位移数据
计算扩散系数
通过这种模拟,可以揭示水分子在不同热力学条件下的扩散特性,为水的物理性质研究提供数据支持。
DFT计算案例:研究分子在催化反应中的作用
在催化剂的研究中,DFT常用于计算反应物与催化剂表面的相互作用以及反应能垒。以氢化反应为例,DFT可以用来研究氢分子在催化剂表面的吸附行为,并计算吸附能。
步骤:
选择适当的催化剂模型(如金属表面)
建立氢分子与催化剂表面的模型
使用DFT方法计算氢分子的吸附能
分析反应的热力学性质,如反应能垒
这种DFT计算可以帮助设计高效的催化剂,优化反应条件,提高催化剂的性能。
总结
分子动力学模拟(MD)和密度泛函理论(DFT)在原理和应用上各有优势。MD模拟更适合描述分子在时间演化过程中的动力学行为,适用于大规模的系统和长时间尺度的研究;而DFT则通过量子力学的框架对分子和固体的电子结构进行精确计算,适用于研究基态能量、分子结构、反应机制等。两者各自的优势和局限性使得它们在不同的应用场景中互为补充,常常结合使用,以获得更加全面的研究结果。
在具体的应用案例中,MD模拟可以用来研究分子的运动行为和物质的宏观性质,而DFT则可以深入分析分子的电子结构和反应性。因此,在多学科领域,MD与DFT的结合为材料科学、催化反应、生物分子研究等提供了强大的计算工具。