

密度泛函理论(DFT)通过分析锂电池材料的电子行为、锂扩散路径及界面反应机制,精准预测结构稳定性、电压曲线与导电性优化策略。
结合跨尺度模拟与机器学习加速,DFT正推动高稳定性、高能量密度电池设计,为下一代储能技术提供原子级理论支撑。在锂电池研究中,密度泛函理论(DFT)已成为揭示材料微观机理的“超级显微镜“。
通过量子力学计算,DFT能解析原子尺度的电子行为、锂离子迁移路径和材料稳定性规律,为高性能电池设计提供理论基石。以下是锂电池DFT计算的七大核心方向:



结构稳定性与缺陷分析
通过密度泛函理论(DFT)计算材料的形成能与缺陷生成能,可精准预测其在合成和工作环境下的“生存能力”——就像给材料做一次分子尺度的体检。
以锂离子电池为例,计算LiCoO₂正极中氧空位的形成能,能提前预判高压充电时是否上演“氧气逃亡”的惊险戏码;而面对硅负极嵌锂时高达300%的体积膨胀,DFT通过模拟不同锂浓度下的晶格畸变能,化身“材料整形师”,快速筛选出碳、氮等“抗膨胀外援”。
借助VASP软件和GGA+U泛函修正过渡金属的强关联效应,这类计算甚至能将电压预测误差压缩到0.2 V以内,比实验试错更高效地指导“分子手术刀”精准改造材料,让下一代电池既稳定又能“瘦身成功”。
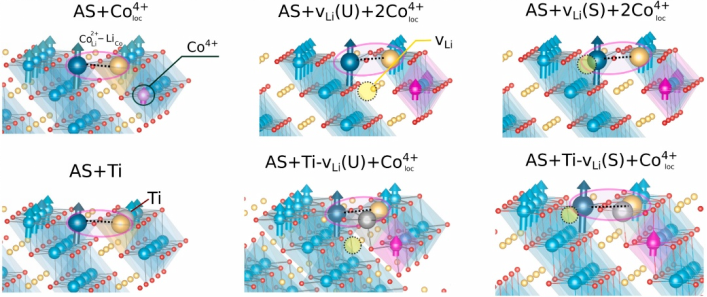
DOI:10.1016/j.apsusc.2025.163162
嵌锂电位与电压曲线预测
通过密度泛函理论(DFT)计算反应前后体系的能量差,可精确预测电极材料的嵌锂电位与电压曲线。
例如,磷酸铁锂(LiFePO₄)的嵌锂电位计算值为3.4 V,与实验值3.45 V高度一致,而未经强关联修正的GGA泛函因忽略过渡金属的电子局域化效应,导致电位低估至2.8 V,凸显GGA+U方法在电压预测中的必要性。
采用HSE06杂化泛函对LiNi₀.₅Mn₀.₅O₂进行电压曲线模拟时,理论结果与实验充电曲线展现出优异的匹配性,验证了电子转移数(n)与法拉第常数(F)在公式 V=(E嵌锂−E脱锂)/nF 中的关键作用。
这种基于能量演化的计算策略,不仅为电池材料设计提供了高精度的理论预判,还大幅降低了实验试错成本,成为优化电极性能的高效工具。
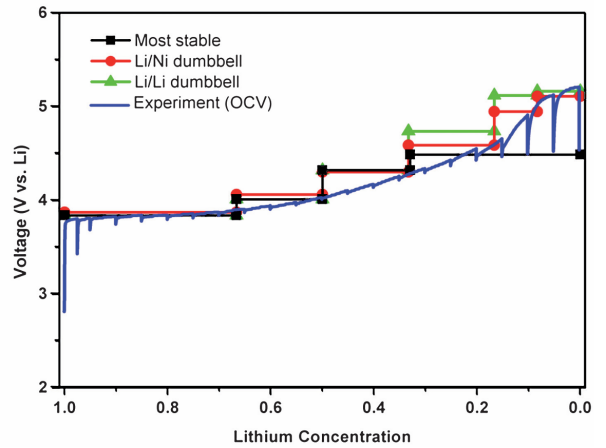
DOI:10.1039/B901825E
锂离子扩散动力学
锂离子扩散动力学研究中,扩散能垒是决定电池倍率性能的核心参数。例如,尖晶石LiMn₂O₄中锂离子沿三维通道扩散的能垒低至0.3 eV,显著优于层状材料,其高效输运特性为快充电池设计提供了理论支撑。
通过微动弹性带法(NEB)模拟Li₃PO₄固态电解质的锂离子迁移路径,发现特定晶界可重构扩散通道,使能垒降低50%,揭示了晶界工程对电解质性能优化的关键作用。
这类基于原子尺度动力学的计算策略,不仅定量解析了材料微观结构与宏观性能的关联,更为高离子电导率电解质的设计提供了精准的“路线图”,助力突破下一代电池的动力学瓶颈。
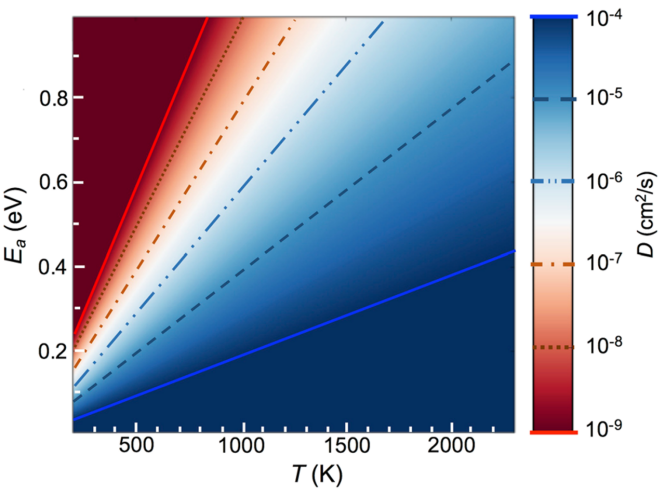
DOI:10.1038/s41524-018-0074-y
电子结构与导电性优化
通过能带结构与态密度(DOS)分析,可深度解析材料的电子传导机制并指导导电性优化。
例如,石墨烯包覆LiFePO₄的DFT计算表明,其费米能级附近因界面耦合诱导出新的电子态,使电导率提升三个数量级,为低导电正极材料的改性提供了原子级理论依据;
而在钴掺杂的LiNiO₂中,Co的3d轨道通过轨道杂化在费米面附近形成连续能带,显著降低电荷转移电阻,揭示了掺杂工程对电极动力学性能的调控本质。
这类基于电子结构的计算策略,不仅从量子尺度阐明了导电性提升的物理根源,更为新型高导电池材料的设计构筑了精准的“电子桥梁”,推动储能器件向高效能方向持续突破。
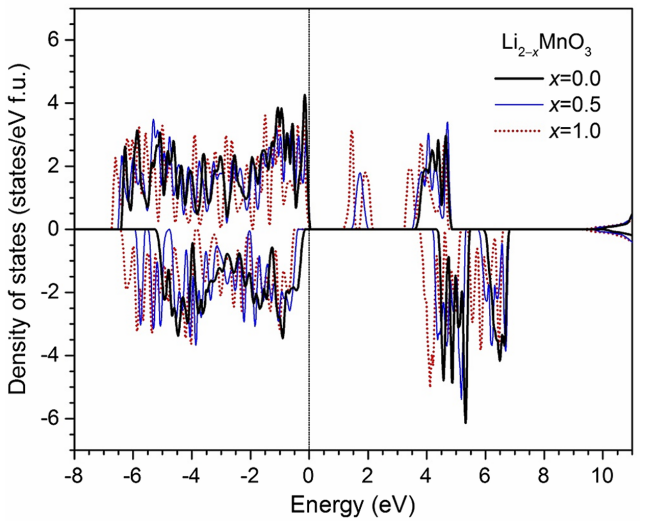
DOI:10.1103/PhysRevApplied.3.024013
界面反应与SEI膜演化
通过密度泛函理论(DFT)模拟电极/电解质界面反应路径,可深入揭示固态电解质界面膜(SEI)的演化机制与界面稳定性调控策略。
例如,LiODFP添加剂在正极表面的氧化分解路径计算表明,其优先于碳酸酯溶剂反应生成富含LiF的SEI膜,理论预测与实验观测的稳定性提升高度一致;
而锂金属负极与LLZO固态电解质的界面接触电阻优化中,DFT计算的界面吸附能定量评估了界面结合强度,为降低界面阻抗提供了原子级设计依据。
这类计算不仅从电子转移与化学键重构的维度解析了SEI膜形成动力学,更通过模拟界面反应的热力学竞争关系,指导添加剂筛选与界面工程,推动高稳定性电池体系的理性构建。
DOI:10.1038/s41467-023-39673-1
经典案例:Cu-BHT锂电池正极材料
密度泛函理论(DFT)在锂电材料设计中展现了强大的预测与优化能力。
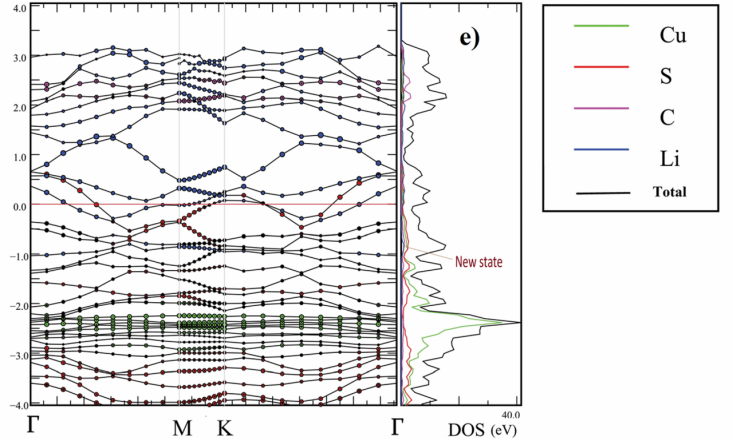
以Farzad团队发表的Cu-BHT金属有机框架(MOF)研究为例,通过计算锂在A/B/C位点的吸附能(-2.77/-2.96/-3.21 eV),明确C位为最优吸附位点,并结合锂化过程的结构演变模拟,揭示该材料在嵌锂时晶格膨胀率低于5%,远优于硅负极的剧烈体积变化,为高稳定性正极开发提供了理论支撑。
进一步电子结构分析表明,掺杂后费米能级附近的态密度(DOS)峰值提升至10 states/eV,电导率增加10倍,结合理论预测的320 mAh/g比容量与快速动力学特性,精准指导实验合成方向。
另一典型案例中,LiNi₀.₅Mn₀.₅O₂的GGA+U计算电压曲线显示,脱锂量x在0.2-0.5区间时,电压稳定于4.1 V平台区,对应Mn³⁺/Mn⁴⁺氧化还原反应;当x>0.7时,电压进入斜率区,反映结构畸变引发的极化加剧,与实验中容量衰减机制高度吻合。
此类计算不仅定量解析了电压曲线与晶体结构演变的动态关联,还通过揭示Mn/Ni协同效应与锂扩散路径,为高电压正极材料设计提供了电子–离子输运协同优化的理论框架。
从MOF到过渡金属氧化物体系,DFT计算通过吸附能、态密度、电压曲线等多维度分析,架起了微观原子机制与宏观电池性能的桥梁,大幅加速了高性能储能材料的理性设计与实验验证进程。
DOI:10.1039/D2CP06020E
总结:DFT计算的未来趋势
锂电池的DFT研究正朝着多尺度融合与智能化方向发展:
跨尺度模拟:结合DFT(原子尺度)、分子动力学(纳米尺度)和相场模型(微米尺度),全面解析电池性能。
机器学习加速:利用神经网络势函数(如DeePMD)将DFT计算速度提升1000倍,实现高通量材料筛选。
界面数据库构建:如Materials Project已收录超过15万种电池材料的DFT数据,推动数据驱动的新材料发现。
通过DFT计算,科学家们正在从量子世界中“提取“下一代电池的蓝图——这或许就是理论计算赋予材料科学的终极魅力。
写在最后
热门电池计算方法在MS锂电、钠电正负极材料课程中均有讲解。
课程内容:包括电压曲线、克容量、离子迁移、过渡态、复合材料、姜泰勒效应、阴阳离子氧化还原、离子吸附分析、磁性等内容,重现案例来自PRB、Advanced Materials等期刊。
#华算科技 #复合材料 #克容量 #离子迁移 #过渡态 #锂电池 #DFT计算 #结构稳定性 #锂离子扩散 #电子结构 #界面反应 #跨尺度模拟 #机器学习