本文主要探讨了吸附能的概念、重要性及应用。吸附能是衡量分子与固体表面相互作用强度的关键指标,其大小受表面材料和分子特性影响。在催化、气体传感、材料设计与环保等领域有重要作用。
在自然界和科技应用中,分子与材料表面之间的相互作用无处不在。我们常常需要了解这些相互作用的强度和稳定性,这就引出了一个非常重要的概念——吸附能。它不仅在基础科学研究中起着关键作用,也在多个技术领域中具有广泛的应用。
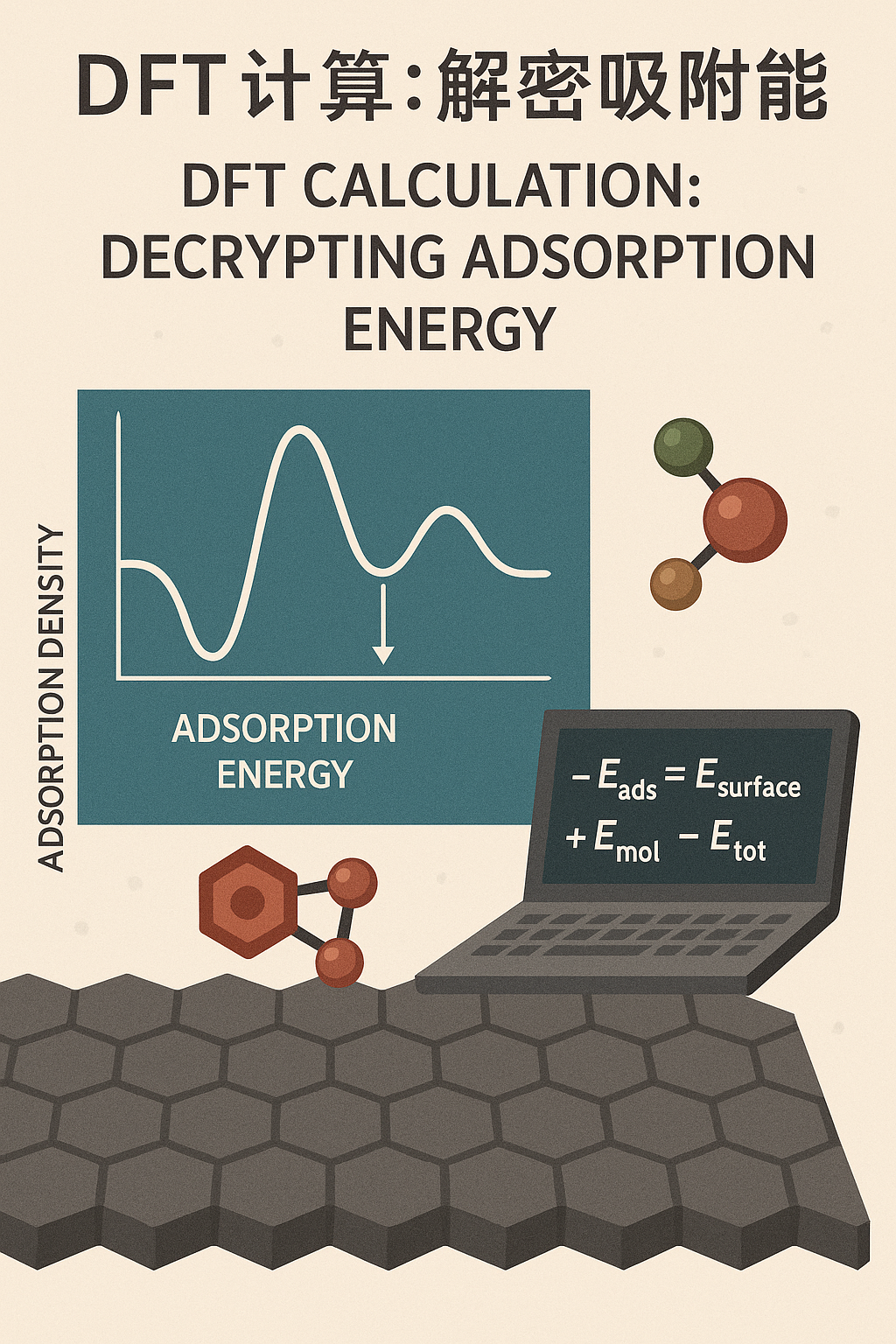
吸附能是指分子(或原子)与固体表面相互作用时,所释放或吸收的能量。简单来说,它是描述分子在表面上停留稳定程度的一个重要指标。当一个分子靠近材料表面并与之发生相互作用时,它会经历一种能量变化过程。
如果吸附能是负的,意味着分子在表面上的吸附是放热的,也就是分子与表面之间有较强的吸引力;如果吸附能是正的,说明吸附过程是吸热的,分子与表面之间的吸引力较弱,吸附的稳定性较差。
吸附能的大小不仅与表面材料的性质有关,还受到分子本身特性的影响。不同的吸附物(如气体分子、离子或有机分子)在不同材料的表面上可能会表现出完全不同的吸附行为。
吸附能是研究和应用中不可忽视的物理量,尤其在以下几个科研方向中,它的作用至关重要:
1. 催化研究: 在催化过程中,反应物首先需要吸附到催化剂表面,然后进行化学反应。吸附能在催化反应中起到了决定性的作用——如果吸附能过强,反应物难以从表面脱附,反应效率低;如果吸附能过弱,反应物则可能无法有效吸附,催化过程无法顺利进行。因此,催化剂的设计通常依赖于对吸附能的精确控制。
2. 气体传感器: 许多气体传感器是基于气体分子与表面材料之间的吸附作用原理。当气体分子吸附到传感器表面时,材料的电学或光学性质发生变化,这种变化可以用来检测气体的存在和浓度。吸附能的研究可以帮助我们选择合适的材料,以优化传感器的灵敏度。
3. 表面工程与材料设计: 吸附能对表面材料的设计和改性至关重要。不同的表面修饰可能会改变吸附能,从而影响材料的性能。例如,在新能源领域,了解电池和燃料电池中分子在电极表面的吸附行为,有助于设计更高效的能源转换设备。
4. 环境保护与污染治理: 吸附技术被广泛应用于水处理、空气净化等领域。通过研究吸附能,我们可以设计更有效的吸附材料,去除空气或水中的污染物。比如,某些重金属离子或有害气体分子会与材料表面发生强烈吸附,利用这一点可以开发高效的净化材料。
文中重点介绍密度泛函理论(DFT)计算对吸附能研究的贡献,通过两个科研实例,展示了DFT在评估不同材料上分子吸附行为及优化催化性能方面的应用,为相关领域研究提供了理论支持与计算方法指导。
对于吸附能的研究,我们通常需要借助计算方法来进行量化,而密度泛函理论(DFT)是目前最常用的工具之一。DFT通过计算电子密度,能够有效地预测分子与固体表面之间的相互作用能,从而精确地计算出吸附能。
通过DFT计算,我们能够获得分子吸附在表面时的能量变化,以及表面与吸附分子之间的电子结构变化。这种计算不仅能告诉我们吸附能的数值,还能揭示吸附过程中的微观机制,例如吸附位点的选择、吸附结构的稳定性等。
DFT的优势在于它的高精度和计算效率,使得它成为分析复杂系统(如催化剂、材料表面、传感器等)中吸附行为的理想工具。通过DFT计算,研究人员可以在没有实验的情况下预测和优化吸附过程,从而为材料设计和技术开发提供理论依据。
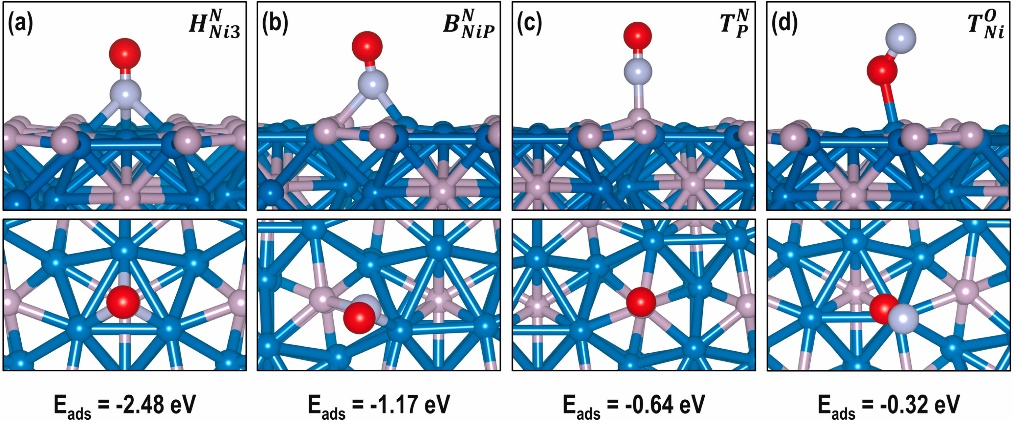
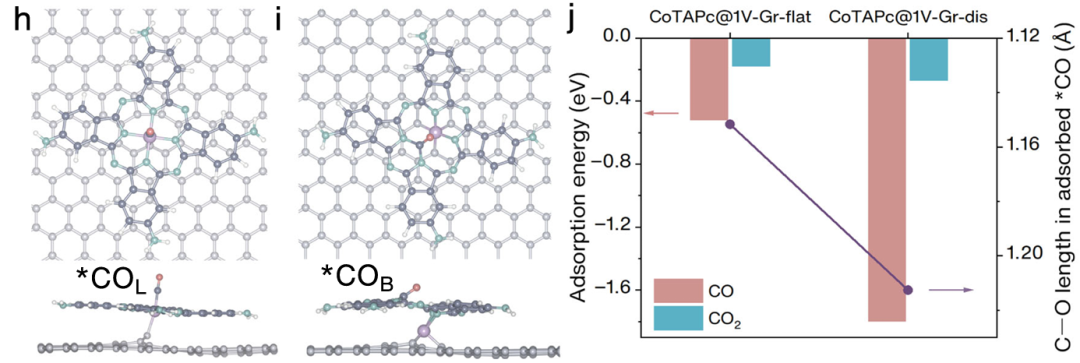
本文通过理论计算与实验结合,揭示了钴酞菁(CoPc)在碳纳米管(CNT)缺陷位上的吸附行为及其在CO₂/CO电还原生成甲醇中的结构演化机制。作者构建了CoPc与CNT单空位相互作用的模型,发现其在电负性电位作用下由近似平面态向离面畸变态转化。
通过DFT计算,两种构型对中间体*CO的吸附能存在显著差异:线性吸附(*CO_L)吸附能为−0.52 eV,而桥式吸附(*CO_B)吸附能高达−1.80 eV,且C–O键长显著延长,显示出更高的反应活性。
吸附能的变化不仅揭示了畸变结构在加氢生成甲醇方面的优势,也解释了电位调控下产物选择性的变化。红外与XAFS实验证实了电位诱导的构型转变及其对吸附模式的影响。
整体而言,吸附能的计算贯穿结构识别、反应路径分析及催化机制构建全过程,是本研究机制解释的核心工具。
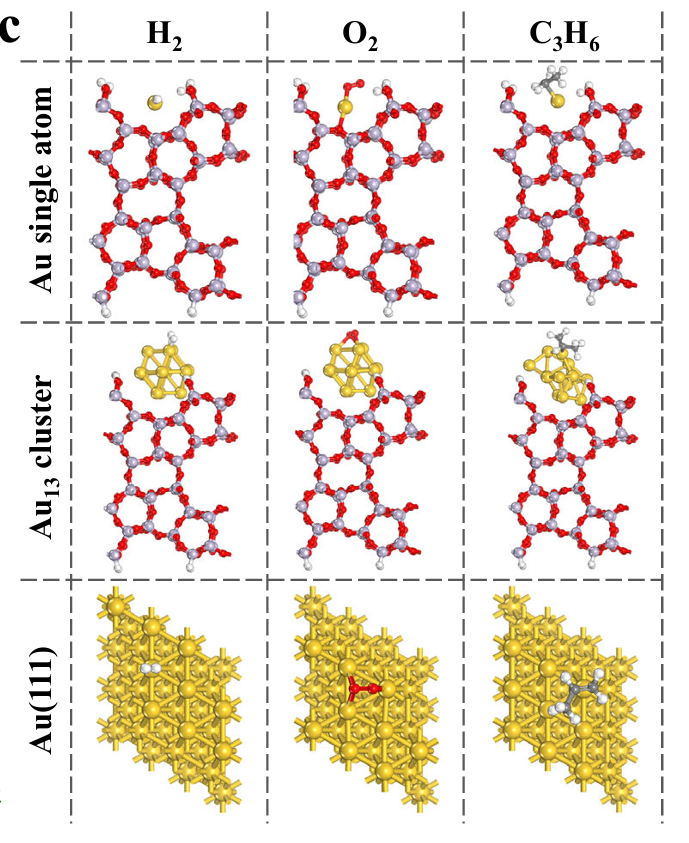
该研究以金单原子催化剂(Au SACs)用于丙烯直接环氧化反应(propylene epoxidation)为核心,重点探讨了其在高反应物覆盖条件下中毒失活的问题,并引入纳米颗粒金(Au NPs)作为“解毒剂”以恢复催化活性。
在这一研究体系中,吸附能的计算在揭示中毒机制、比较不同尺度催化剂反应性以及构建催化协同机制中起到了核心作用。
作者利用DFT方法系统比较了三种金物种(Au 单原子、Au₁₃团簇和Au(111)面)对反应物(H₂、O₂、C₃H₆)的吸附行为,并明确其反应路径。吸附能的计算显示,C₃H₆在Au SACs上的吸附能显著高于在Au(111)上的值,体现出更强的吸附倾向。
该结果虽然从催化角度体现了Au SACs对C₃H₆具有优异的活化能力,但也揭示出其在高覆盖状态下易被C₃H₆“淹没”,导致H₂和O₂无法竞争性吸附,从而阻断了关键的过氧中间体(OOH·)的生成链路。
在进一步的反应路径能垒分析中,Au SACs虽然在生成环氧丙烯的路径上具有最低能垒(0.20 eV),但该优越性受限于其强吸附C₃H₆所带来的中毒效应。
为验证该结论,研究者结合分子动力学模拟,观察到在反应温度下C₃H₆分子在Au SACs表面高度富集,而H₂几乎难以吸附。这一动态吸附过程通过吸附能计算得到实验证实,并进一步通过径向分布函数确认C₃H₆的主导吸附地位。
相较之下,Au NPs显示出对C₃H₆和H₂的中等吸附能力,表现为更平衡的吸附行为。这使得其不仅能生成OOH·自由基,还能通过解吸作用将中间体转移至Au SACs上,使得被C₃H₆覆盖的Au SACs活性位点得以重新激活。
吸附能的协同调节成为Au SACs与Au NPs之间实现反应物覆盖匹配和协同催化的关键机制。
综上所述,吸附能的计算在本研究中不仅揭示了催化剂中毒机制和尺寸效应,也提供了优化催化性能、构建协同催化策略的理论基础。它在链接电子结构与宏观反应性能方面发挥了关键作用,是整篇研究的理论支柱。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!