

本文深入探讨催化领域中吸附与活化的关键指标,阐明其物理意义,并详细阐述密度泛函理论(DFT)在这些指标的预测与分析中的核心作用。催化过程的效率和选择性在很大程度上取决于反应物在催化剂表面的吸附和随后的活化。
本文详细介绍了吸附能、d带中心、电荷转移等吸附指标,以及活化能、过渡态和反应路径等活化指标的定义、计算方法和物理化学意义。特别地,DFT作为一种强大的计算工具,能够提供原子尺度的电子结构和反应机制洞察,从而预测催化剂的活性、选择性和稳定性。
催化过程中的吸附与活化
催化剂性能的核心在于其对反应物的吸附能力以及随后活化这些吸附物以促进化学转化的能力。反应物与催化剂表面结合的强度需适中,既要足够强以促进反应,又不能过强导致催化剂失活,即所谓的“中毒”现象。这种结合强度的平衡是实现高效催化的关键。
在催化研究中,密度泛函理论(DFT),已成为主流研究工具。DFT能够提供实验难以或无法获得的原子级洞察,例如详细的电子结构和反应机制信息。这种能力使得研究人员能够更深入地理解催化过程,从而加速开发更高效、更具选择性的催化剂。
DFT在催化剂设计中促进了从传统的“试错法”向“理性策略”的转变。如果DFT能够在实验之前预测催化剂的性质和反应机制,那么它就能显著减少所需的物理实验次数,这直接导致了更高效和理性的设计过程,从而影响催化剂开发的成本和速度。
吸附的关键指标与物理意义
吸附能
吸附能(Eads)量化了分子与表面结合的强度。它是吸附物(分子)与基底(表面)之间相互作用强度的度量。如果吸附能定义了分子结合的强度,那么它就决定了反应物在表面上的可用性和产物脱附的难易程度。
这直接影响了反应的整体能量图景,从而影响其速度(动力学)和平衡(热力学)。例如,研究发现,所有反应中间体的吸附能集合完全定义了反应的热力学。
DFT计算吸附能的公式为:Eads=Eslab/adsorbate−Eslab−Eadsorbate,其中Eslab/adsorbate是结合态的体系总能量, Eslab是孤立的基底总能量, Eadsorbate是孤立的吸附分子总能量。
准确预测吸附能对于设计具有高选择性、稳定性和活性的催化剂至关重要。如果吸附能决定了结合强度,那么控制它就可以精确调整反应物在表面停留的时间、转化的难易程度以及产物脱离的容易程度。这直接转化为催化剂性能(活性、选择性)的优化,并防止强结合导致中毒等问题(稳定性)。
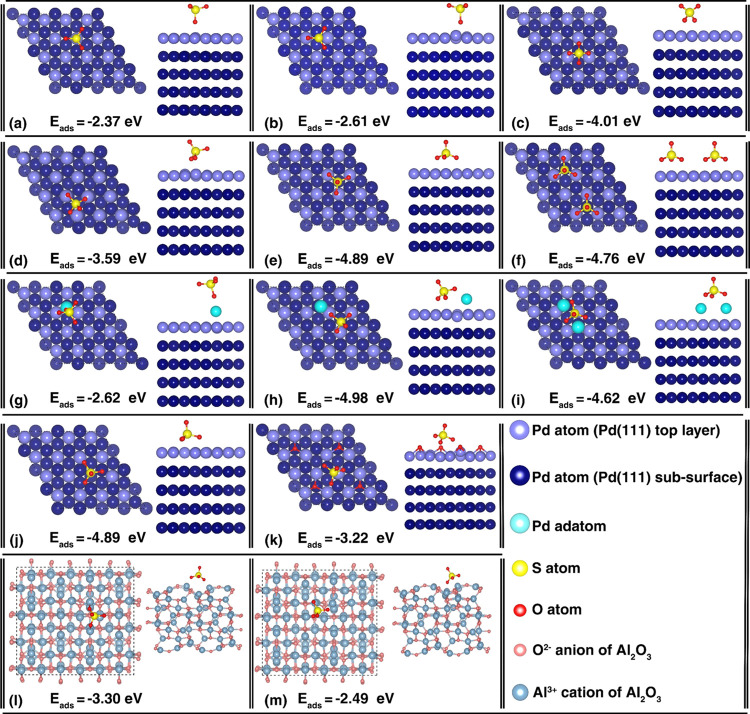

DOI: 10.1021/jacs.2c03088
物理吸附与化学吸附的区分
吸附过程可分为物理吸附(Physisorption)和化学吸附(Chemisorption),它们在键合性质、能量、温度、层形成、可逆性和选择性方面存在显著差异。
物理吸附涉及弱的范德华力,能量较低,通常发生在低温下,接近或低于吸附物的沸点。它可以形成多层吸附,通常是可逆的,并取决于压力和温度,且是非选择性的。
相比之下,化学吸附涉及吸附物与表面之间形成强的化学键,能量较高,并需要特定的活化能。它发生在较高温度下,超过吸附物的沸点,仅限于单分子层,通常是不可逆的或不易逆的,且是选择性的,需要形成化学键。
DFT可以通过计算出的吸附能范围和键合性质来定量区分物理吸附和化学吸附。在势能曲线中,化学吸附会在较短距离处出现更深的能量最小值,表明更强的结合,而物理吸附则由弱范德华力引起。DFT泛函正在发展以准确捕捉强的共价/离子键合(化学吸附)和范德华相互作用(物理吸附)。
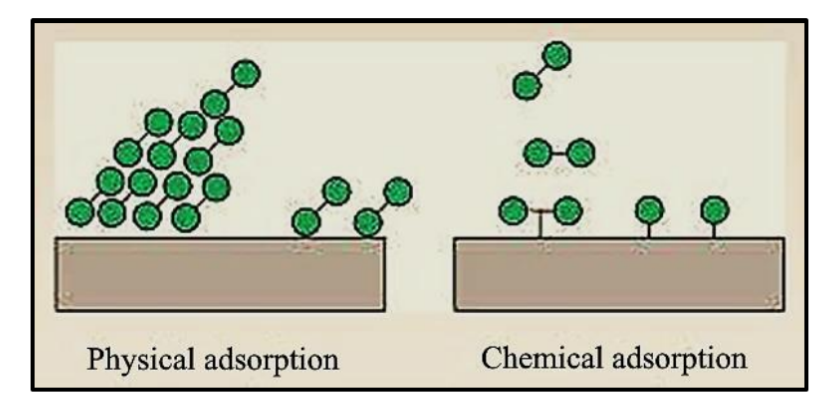
DOI:10.37628/IJTCK
物理吸附与化学吸附的对比

电子结构指标
d带中心理论
d带中心(εd)是吸附物与过渡金属表面之间键合强度的良好初步指标。d态能量相对于费米能级越高,反键态能量越高,吸附键越强。该模型已通过实验验证,并广泛用于关联不同金属表面上各种分子的吸附能。
d带中心位置直接影响表面反应性和吸附强度。更高的d带中心导致更强的键合,并且表面反应性随晶格膨胀而增加。这建立了一个清晰的因果关系:电子结构(d带中心)影响键合强度,进而影响表面反应性。
d带模型为预测不同过渡金属的催化活性趋势提供了强大的描述符,从而实现理性催化剂设计。如果一个单一的电子参数(d带中心)可以预测吸附强度的趋势,那么它就成为筛选和设计新催化剂的宝贵“描述符”,而无需计算每一个吸附能。
例如,计算出的CO和O吸附能与εd相关联,证明了其预测能力。DFT计算中调制d带中心位置的策略已被用于设计高效金属催化剂。
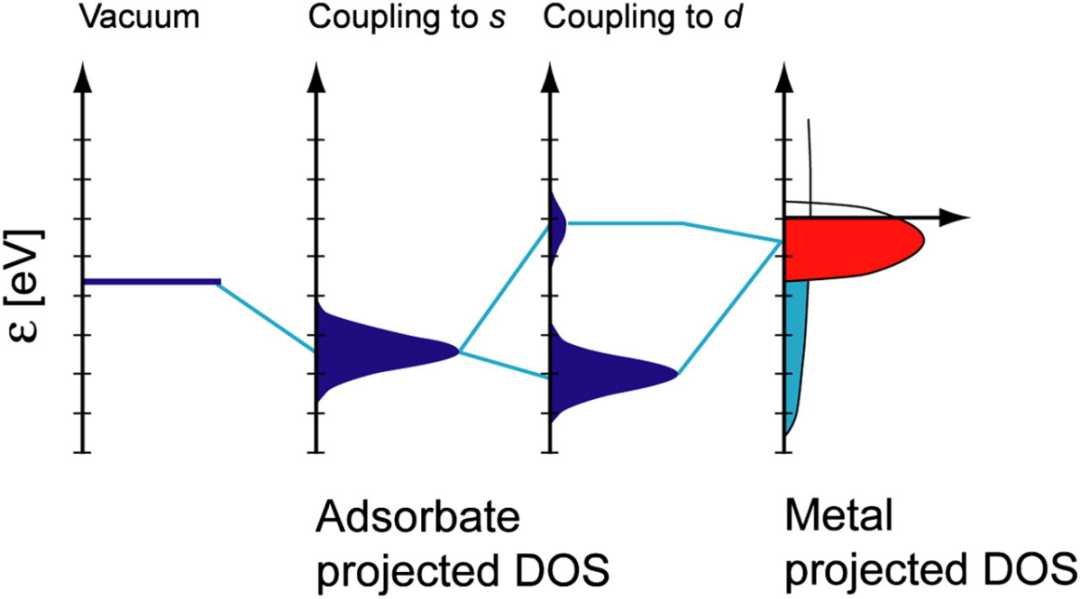
DOI:10.1073/pnas.1006652108
电荷转移
吸附物与金属表面之间的电荷转移是许多气-固反应(包括非均相催化)中的关键步骤。DFT计算可用于分析和量化这种电荷转移。它影响界面处的电子能级,并可能影响金属表面的功函数。电荷转移的程度和方向会随吸附物取向以及分子和基底的固有性质而变化。
电荷转移直接影响键合形成以及吸附物和催化剂的电子状态,从而影响吸附强度和反应性。电荷重新分布伴随着界面电子能级的重新排列。吸附物更高的极化性(可通过电荷转移引起的表面极化诱导)导致更好的吸附。这些现象表明电荷转移是化学键形成背后的基本电子事件。
通过DFT理解电荷转移机制对于通过操纵活性位点处的电子环境来调节催化剂性能(如活性和选择性)至关重要。
如果电荷转移可以通过分子排列或催化剂组成(例如,CeOx/Ag(111)中Ag作为电子供体给CeOx,促进CO2吸附和还原)进行调控,那么控制这种现象就可以有目的地设计催化剂。与物理和化学性质相关的“界面描述符”对于理解和设计金属负载催化剂至关重要。
活化的关键指标
过渡态
过渡态(TS)是反应坐标上反应物和产物之间的一种暂时、不稳定、高能量的状态。它是势能面(PES)上的一个鞍点,表示在一个方向(反应坐标)上是最大值,而在所有其他方向上是最小值。虽然无法直接观察到,但其性质(几何结构、能量、振动频率)可以通过DFT等计算方法推断。
过渡态的性质(几何结构、能量)决定了活化能,从而决定了反应速率。过渡态被定义为“反应坐标上的最高能量状态”,这直接将其能量与活化能联系起来。过渡态的几何结构决定了在能量峰值处发生的具体相互作用和键重排,从而影响反应路径。
理解和稳定过渡态是催化剂设计的关键策略,因为它直接导致活化能降低和反应速率提高。过渡态理论(TS)为理解催化剂如何降低活化能和提高反应速率提供了基础,并通过关注过渡态的稳定化来帮助设计和优化新催化剂。
DFT过渡态计算可以预测选择性,即使是微小的能量差异也至关重要,并且机器学习可以增强这些预测。
反应路径与中间体
催化反应通过一系列基元步骤进行,形成各种瞬态中间体和反应路径。DFT是研究这些反应路径、识别中间体和确定速率决定步骤(RDS)的强大工具。这些中间体的吸附能与反应物或产物的吸附能一样重要,共同定义了反应的能量学。
反应中间体和过渡态沿路径的稳定性和能量决定了整体反应机制和动力学。与反应中间体对应的吸附能必须包含在内,才能完全定义特定反应路径的反应能量学。势能面(由DFT计算)用于识别过渡态并理解反应机制。
DFT识别中间体和速率决定步骤的例子直接将其与催化活性联系起来。DFT揭示原子级详细反应机制的能力是理性催化剂设计的基础,使研究人员能够优化条件以获得所需产物并避免不必要的副反应。
如果DFT能够描绘出完全不同的反应路径并识别最可能的反应路径,它就能让化学家理解反应为何以某种方式进行。这种理解随后被用于指导新催化剂的设计以提高性能和通过加速特定步骤来适当控制选择性。反应路径描述符对于催化剂设计至关重要。
DOI:10.1002/anie.20250809
DFT方法在吸附与活化预测
DFT基本原理简述:Hohenberg-Kohn定理与Kohn-Sham方程
密度泛函理论(DFT)是一种量子力学方法,主要研究多电子体系的电子结构,特别是其基态。它基于Hohenberg-Kohn定理,该定理指出体系的基态电子密度唯一地决定了体系的所有基态性质。
Kohn-Sham方程通过引入一个非相互作用的参考体系,提供了一种计算该密度和能量的实用方法。总能量表示为其密度的泛函。与其他量子化学方法相比,DFT在精度和计算成本之间提供了最佳折衷。
DFT的核心优势在于将复杂的多电子问题简化为更易于处理的电子密度问题,使其在计算上对于催化相关的大型体系变得可行。DFT的计算效率(“精度和计算成本之间的最佳折衷”,“比Hartree-Fock方法更具通用性”)直接归因于Hohenberg-Kohn定理和Kohn-Sham假说,它们将复杂性从波函数降低到电子密度。这是理解DFT为何被广泛采用的基础。
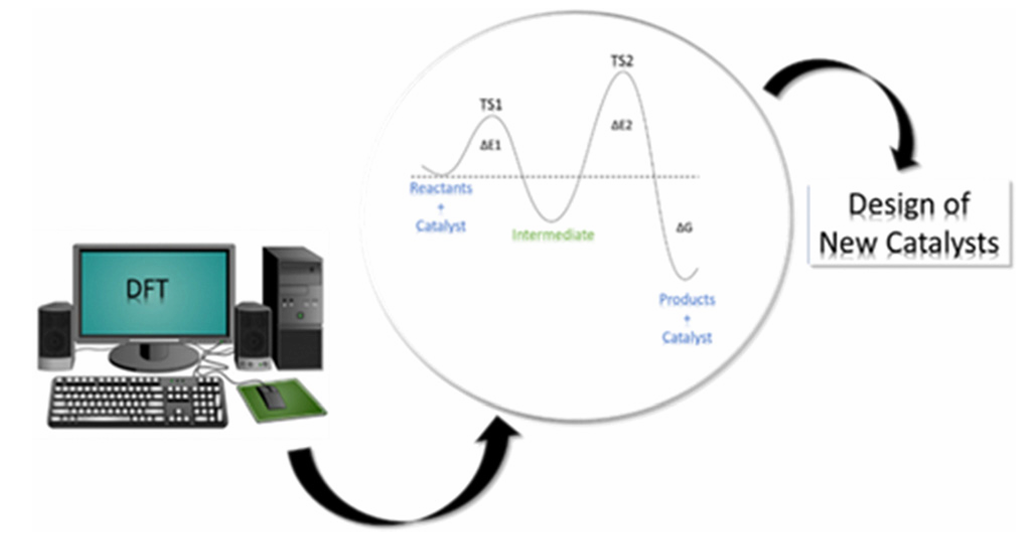
DOI: 10.1039/d4cp00266k
吸附能的DFT计算:模型构建、泛函选择与色散校正
在DFT吸附研究中,模型通常由基底材料的平板和其上的吸附分子组成。平板应足够厚以代表体相性质,并在表面平面上具有周期性边界条件。吸附分子应放置在合理的构型中。由于计算成本,通常使用理想化模型,如金属表面、扩展金属氧化物表面或纳米团簇。
DFT结果的准确性在很大程度上取决于所选择的交换-关联泛函。常见的类型包括局域密度近似(LDA)、广义梯度近似(GGA)(例如PBE、BLYP)和杂化泛函。杂化泛函通常提供更高的准确性,但计算成本更高。
传统的DFT泛函(特别是局域/半局域泛函)可能无法准确捕捉长程范德华(vdW)相互作用,这对于准确计算吸附能至关重要,尤其对于物理吸附或大型分子体系。包含色散校正(例如DFT-D3、vdW-DF)显著提高了金属表面吸附能的准确性。
DFT的“最佳折衷”伴随着选择合适泛函和考虑色散力的挑战,这对于准确的吸附能计算至关重要。目前没有单一的DFT交换-关联泛函能够同时准确预测过渡金属上由强共价或离子键合主导的吸附能,以及由范德华相互作用贡献很大的吸附能。这指出了一个重要的方法学细微之处和当前的研究领域(开发更好的泛函)。
计算参数(模型、泛函、色散校正)的仔细选择对于DFT在催化中的可靠性和预测能力至关重要。如果所得结果的可靠性在很大程度上取决于所选方法和模型中涉及的近似,那么用户需要理解DFT并非一个“黑箱”解决方案。对特定泛函和色散校正的讨论为获得准确结果提供了可操作的知识。
活化能与反应路径的DFT计算:过渡态搜索、势能面分析与过渡态理论 (TST)
DFT用于在势能面(PES)上定位过渡态。这涉及识别PES上的鞍点,它们代表反应坐标上的最高能量点。PES描述了反应的能量景观,允许识别反应路径、中间体和过渡态。DFT计算能够绘制这些复杂的能量景观。
过渡态理论(TST)是分析非均相催化反应机制和动力学的基本框架。它假设反应速率由反应物通过过渡态的速率决定。DFT衍生的反应物、产物和过渡态的能量和振动频率被用作TST的输入,以计算速率常数和反应速率。
利用DFT准确识别过渡态和绘制PES是计算可靠活化能和理解反应动力学的直接先决条件。过渡态被明确指出是“反应坐标上的最高能量状态”,并且TST根据通过此状态的速率来计算速率。
因此,通过DFT准确找到并表征过渡态是动力学预测的基础。DFT为微观动力学建模提供了必要的“速率常数”和“活化能”。DFT结合TST提供了一个强大的预测框架,通过识别速率决定步骤和阐明竞争反应路径,预测催化剂的活性和选择性。
如果DFT+TST能够识别最慢的步骤(速率决定步骤)并比较不同路径的能量,它就能让研究人员理解为什么一种产物比另一种更受青睐。这直接转化为设计催化剂以促进所需路径或抑制不期望路径,从而控制选择性。然而,由于微小的能量差异和构象采样,预测选择性仍然具有挑战性。
预测催化剂性能:活性、选择性与稳定性
DFT为预测催化剂性能的三个关键方面:活性、选择性和稳定性,提供了一个全面的计算框架。
活性: DFT通过计算反应物/中间体的吸附能和基元步骤的活化能来预测催化剂活性。较低的活化能垒通常意味着更高的活性。DFT可以识别活性位点并预测催化剂如何与反应物相互作用。
选择性: DFT通过分析竞争反应路径及其相对活化能垒来帮助预测选择性。通过比较不同产物过渡态的能量,DFT可以确定哪条路径在动力学上更有利。催化剂结构可以设计成加速导致目标产物的特定步骤,从而控制选择性。
稳定性: DFT有助于理解和预测催化剂稳定性,这受到烧结、中毒和积碳等失活机制的影响。
烧结 (Sintering): 高温导致活性颗粒团聚,减少表面积。DFT可以模拟表面能和重构,这些与抗烧结性有关。例如,DFT研究了δ-MoC表面在不同碳化条件下的稳定性和形貌。
中毒 (Poisoning): 杂质强烈吸附到活性位点,使其不可用。DFT可以模拟潜在毒物(例如H2S形成Cu-S键)的吸附强度及其对活性位点可用性和电子结构的影响 。
积碳 (Coking): 碳质沉积物堆积堵塞活性位点和孔隙。DFT可以研究积碳形成路径以及不同催化剂表面对积碳的敏感性。例如,Ni(111)表面由于较低的C-O键断裂活性,比阶梯状Ni(211)表面更不易被积碳失活。
催化剂失活机制(烧结、中毒、积碳)与表面相互作用和电子性质密切相关,这些都可以通过DFT进行建模。中毒涉及杂质粘附到活性位点,积碳是碳堆积,烧结是颗粒团聚。
DFT能够模拟吸附能(用于中毒)、反应路径(用于积碳)和表面稳定性/重构(用于烧结),这直接将这些宏观失活现象与原子级相互作用联系起来。H2S形成Cu-S键的化学中毒可以通过DFT进行建模。
总结
本文详细阐述了催化领域中吸附和活化的关键指标及其物理意义,并深入探讨了密度泛函理论(DFT)在这些指标的预测和分析中的核心作用。吸附能、d带中心、电荷转移和振动频率作为吸附的关键指标,提供了分子与催化剂表面相互作用强度和性质的原子级信息。活化能、过渡态和反应路径则揭示了反应动力学和机制的本质。
DFT作为一种强大的第一性原理计算工具,通过其对电子结构的精确描述,能够定量预测这些关键指标。它弥合了微观原子相互作用与宏观催化性能之间的鸿沟,使得研究人员能够从根本上理解催化剂的活性、选择性和稳定性。
通过DFT,可以识别最佳吸附位点、阐明速率决定步骤、探索竞争反应路径,并预测催化剂在烧结、中毒和积碳等失活机制下的行为。