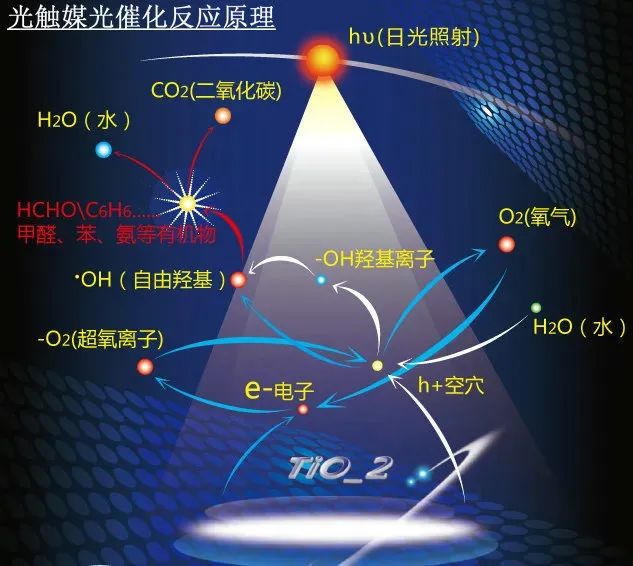
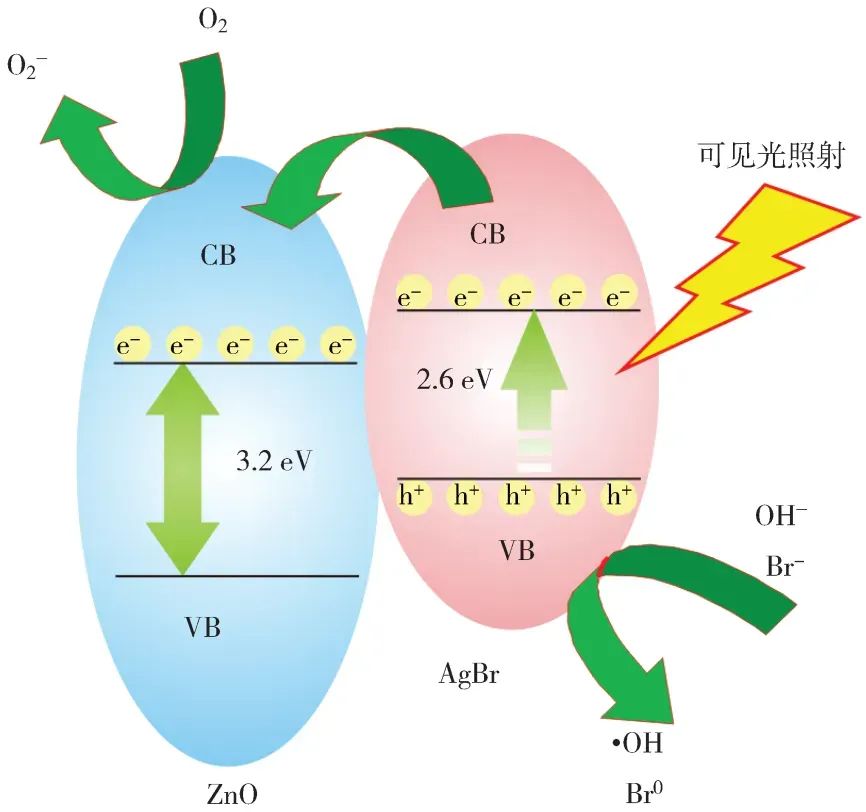
光催化作为一种将太阳能转化为化学能或热能的重要技术,近年来在能源和环境领域得到了广泛关注。为了深入理解光催化材料的结构-性能关系,并优化其性能,理论计算在光催化研究中扮演着至关重要的角色。
本文华算科技将从多个角度详细探讨光催化可以进行的理论计算,包括电子结构与能带特性、光学与载流子输运特性、表面反应与界面调控、稳定性与材料设计等方面,并结合具体的研究案例和方法,展示理论计算在光催化中的应用。
光催化材料的电子结构决定了其光吸收性能和载流子分离能力。通过密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)等方法,可以精确计算材料的能带结构、态密度(DOS)和光吸收特性。
研究表明,氮掺杂的TiO₂(TiO₂₋ₓNₓ)在可见光下表现出优异的光催化活性,这主要归因于其能带隙的缩小和光吸收能力的增强。通过第一性原理计算,可以揭示氮掺杂对TiO₂能带结构的影响机制,从而为设计高效可见光响应的光催化剂提供理论指导。
此外,层状钙钛矿材料(如Ba₂Bi₃Nb₂O₁₁I)的高极化率碘离子可以显著改变其能带结构,从而延长载流子寿命,提高光催化效率。通过DFT计算,可以分析碘离子对钙钛矿材料的电子结构和光学性质的影响,为开发新型光催化剂提供理论支持。
光催化材料的光学性能和载流子输运特性是影响其光催化效率的关键因素。通过TD-DFT和广义梯度近似(GGA)等方法,可以模拟材料在不同波长下的光吸收特性,并预测其光响应范围。
研究表明,CdS@Au/MXene复合材料的优异光催化性能与其界面处形成的“双肖特基势垒”密切相关。通过DFT计算,可以分析CdS纳米颗粒与Au纳米颗粒之间的电荷转移机制,从而揭示其高效光催化性能的内在原因。
此外,载流子的输运特性对光催化反应的效率也有重要影响。通过分子动力学模拟和第一性原理计算,可以研究载流子在材料中的扩散路径和复合机制。例如,CdS中Cd空位的引入可以产生内建电场,促进载流子的定向传输,从而提高光催化效率。通过DFT计算,可以分析Cd空位对CdS能带结构和载流子输运特性的影响,为优化光催化材料提供理论依据。
光催化反应的效率高度依赖于反应物在催化剂表面的吸附行为和后续的反应路径。通过DFT计算,可以精确模拟分子或原子在材料表面的吸附构型、吸附能以及电子结构变化。
在析氢反应(HER)中,反应中间体的吸附自由能(ΔG*)是评估催化剂活性的关键描述符。通过DFT计算,可以分析不同催化剂表面的吸附自由能,从而筛选出具有高活性的催化剂。
此外,过渡态理论(TST)和爬坡弹性带(NEB)方法可用于确定反应路径上的能垒,从而识别决速步骤。在水氧化反应中,双金属MOFs(如Fe-MOFs)可以显著降低N₂的活化势垒,从而提高光催化氨合成的效率。通过DFT计算,可以分析双金属节点的电子结构和反应路径,从而揭示其高效催化性能的机理。
光催化材料的稳定性是其实际应用的关键因素之一。通过第一性原理计算和分子动力学模拟,可以研究材料在不同环境下的稳定性,并预测其长期性能。
研究表明,g-C₃N₄及其改性材料在光催化降解有机污染物和CO₂还原方面表现出优异的性能。通过DFT计算,可以分析g-C₃N₄的电子结构和电荷转移特性,从而理解其高催化活性的机理。
此外,机器学习辅助设计方法可以结合高通量计算和自动化实验,实现材料研发的闭环优化。通过主动学习策略,可以迭代地选择最具信息量的数据点进行计算或实验验证,从而以最少的资源消耗优化模型性能。这种方法不仅可以显著缩短研发周期,还能降低成本,为光催化材料的开发提供新的思路。
理论计算在揭示光催化反应机理方面也发挥了重要作用。例如,研究表明,水-甲醇团簇在金红石TiO₂表面的协同作用对光催化反应速率有重要影响。通过Operando NMR表征、DFT计算和AIMD计算相结合的方法,可以揭示水分子和甲醇分子在TiO₂表面的吸附行为和反应路径,从而理解其光催化反应的机理。
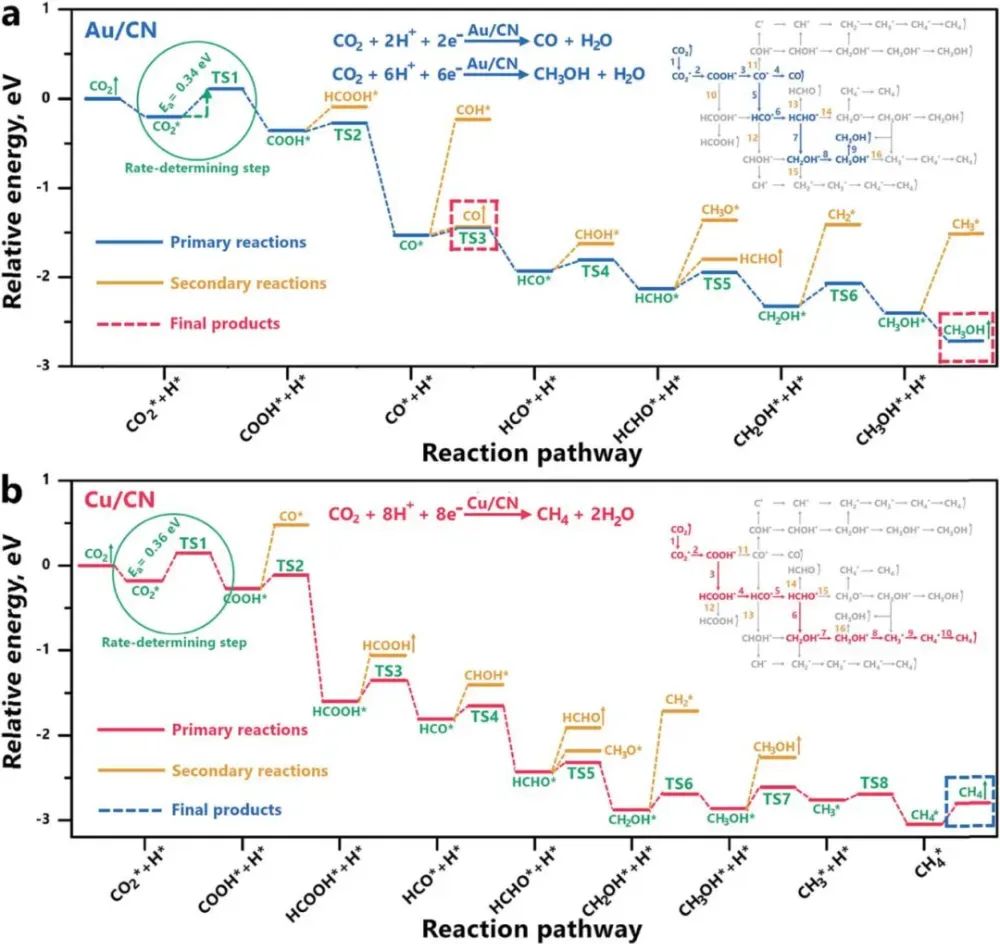
此外,光催化CO₂还原成C₂+产物的反应机理也得到了深入研究。通过DFT计算,可以分析CO₂分子在催化剂表面的吸附和活化过程,从而揭示其反应路径和能垒。研究表明,CoOx量子点修饰的多孔石墨烯在光催化CO₂还原中表现出优异的性能,这主要归因于其高效的N₂解离和电子转移能力。
理论计算在光催化材料的结构设计中也发挥了重要作用。例如,研究表明,通过调控材料的晶体结构和表面微观结构,可以显著提高其光催化性能。例如,g-C₃N₄的结构调控可以通过热聚合法、水热法和溶胶-凝胶法等多种方法实现,并通过DFT计算分析其电子结构和电荷转移特性,从而理解其高催化活性的机理。
此外,界面调控也是提高光催化性能的重要手段。例如,通过在半导体材料表面负载双功能助催化剂(如Rh₀-Rh³+位点),可以显著提高其光催化水氧化和还原反应的效率。通过DFT计算,可以分析助催化剂与半导体材料之间的相互作用,从而揭示其高效催化性能的机理。
理论计算在光催化材料的性能优化中也发挥了重要作用。研究表明,通过调控材料的掺杂和缺陷结构,可以显著提高其光催化性能。CdS中Cd空位的引入可以产生内建电场,促进载流子的定向传输,从而提高光催化效率。通过DFT计算,可以分析Cd空位对CdS能带结构和载流子输运特性的影响,从而为优化光催化材料提供理论依据。
此外,通过调控材料的能带结构和表面性质,可以显著提高其光催化性能。研究表明,通过引入“双肖特基势垒”结构,可以显著提高CdS@Au/MXene复合材料的光催化性能。通过DFT计算,可以分析CdS纳米颗粒与Au纳米颗粒之间的电荷转移机制,从而揭示其高效光催化性能的内在原因。
理论计算的结果需要通过实验验证来确认其可行性。研究表明,通过水热法成功制备了具有Cd空位缺陷的Cd₁₋ₓS,并通过XRD、HRTEM、EPR、光催化性能、紫外-可见漫反射光谱、XPS、PL等表征方法验证了其光催化性能。通过DFT计算,可以进一步分析Cd空位对CdS光催化性能的影响,从而为实验结果提供理论支持。
通过实验验证理论计算的结果,可以进一步优化光催化材料的性能。研究表明,通过引入“双肖特基势垒”结构,可以显著提高CdS@Au/MXene复合材料的光催化性能。通过实验验证,可以进一步优化其结构和性能,从而为实际应用提供理论支持。
随着理论计算方法的不断发展,光催化材料的研究将更加深入。例如,通过结合高通量计算和机器学习方法,可以实现材料研发的闭环优化,从而显著缩短研发周期并降低成本。此外,通过结合实验研究和理论计算,可以进一步揭示光催化反应的机理,并优化其性能。
此外,光催化材料的未来发展方向还包括开发新型光催化剂、提高光催化效率和稳定性、拓展光催化应用领域等。通过理论计算,可以为这些目标提供理论支持和指导,从而推动光催化技术的进一步发展。
理论计算在光催化研究中发挥着重要作用,涵盖了从电子结构、光学性能、表面反应到材料设计等多个方面。通过DFT、TD-DFT、TST、NEB等方法,可以深入理解光催化材料的结构-性能关系,并优化其性能。此外,结合实验验证和机器学习方法,可以实现材料研发的闭环优化,从而显著提高光催化材料的性能和应用前景。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!