本文深入探讨了离子扩散在电池材料中的关键作用,阐释了其对电池充放电速率、循环寿命及整体性能的显著影响。文章着重讲述了计算离子扩散系数的重要性,指出其对电池设计优化和性能预测具有不可替代的指导价值。
在方法层面,详细介绍了基于密度泛函理论(DFT)和分子动力学(MD)的两种计算离子扩散系数的主要方法。DFT方法以其精确性,被广泛应用于固体电极材料中离子迁移能垒的计算,从而有效预测离子在电极材料中的扩散效率,并为材料筛选与优化提供理论支持。
而MD方法则以其对复杂体系的模拟能力,被用于计算电解液或聚合物电解质中的离子扩散系数,能够深入研究不同电解液在多样化条件下的性能表现,进而为电池的进一步优化提供有力的理论依据。
离子扩散是指电池中离子(如锂离子、钠离子等)在电极材料和电解液之间移动的过程。在电池充放电过程中,离子通过电解液迁移并与电极反应,完成电池的能量储存和释放。
离子扩散速率直接影响电池的充放电速率、循环寿命及其整体性能。提升离子扩散性能,特别是在电极材料中的扩散能力,是提升电池性能的重要途径。
在电池设计中,理解并优化离子扩散系数对于提高电池性能至关重要。扩散系数决定了离子在电池内部移动的速度和效率,进而影响到电池的充电速度、使用寿命以及安全性。
例如,高扩散系数有助于提高电池的功率密度和循环稳定性,而低扩散系数则可能导致电池在快速充放电过程中的性能下降。
通过计算离子扩散系数,研究人员可以预测不同电池材料在实际工作中的表现,并在此基础上优化电池设计。此外,合理的离子扩散模型能帮助开发出适合不同工作环境(如低温或高温)的电池。
离子扩散系数的计算方法主要有两种:基于密度泛函理论(DFT)和分子动力学(MD)的计算。
密度泛函理论(DFT)通常用于计算固体电极材料中离子的迁移能垒。迁移能垒是指离子从一个位置迁移到另一个位置所需克服的能量障碍。在电池电极中,离子的迁移能垒是影响离子扩散速率的关键因素之一。
DFT能够精确计算材料中的离子迁移能垒,从而预测离子在电极材料中的扩散效率。通过DFT的计算,可以评估不同材料的离子扩散性能,并对材料进行筛选或优化。
通过DFT计算,研究人员能够精确计算锌离子在NVO和INVO晶格中的吸附能及扩散路径,从而揭示了掺碘如何有效降低锌离子的扩散能垒。
在图4A中,DFT计算显示,掺碘后的INVO材料相比于NVO,锌离子在最稳定吸附位点的吸附能明显降低,从-1.50 eV降至-1.44 eV。这一变化是由于碘原子的较大原子半径和较低的电负性,导致晶格的膨胀和锌离子与材料之间的吸引力减弱。
接着,DFT计算还对不同的扩散路径进行了分析,图5A至5D展示了锌离子在NVO和INVO中的四条主要扩散路径及其迁移能垒。
在NVO中,最具挑战性的扩散路径是锌离子沿层间空间的扩散,其能垒高达1.22 eV,而在INVO中,这一扩散路径的能垒显著降低至0.99 eV,意味着锌离子可以更加容易地穿过材料的晶格结构。
由此可见,碘的引入不仅有效扩展了晶格结构,还通过降低迁移能垒来增强了锌离子的扩散能力,进而提高了材料的电化学性能。通过这些计算,DFT理论为研究者提供了一个理论框架,帮助他们理解并优化电池材料中的离子扩散行为,这对于开发高性能的电池材料至关重要。
分子动力学(MD)方法则用于计算电解液或聚合物电解质中的离子扩散系数。通过模拟离子在溶剂中的运动,MD可以计算离子在电解液中的扩散行为及其系数。MD方法可以模拟更复杂的体系,如电解液中溶剂和盐的交互作用,从而得到更加全面的扩散系数数据。
通过MD模拟,可以研究不同电解液在不同温度、压力等条件下的性能,并为电池的优化提供理论支持。
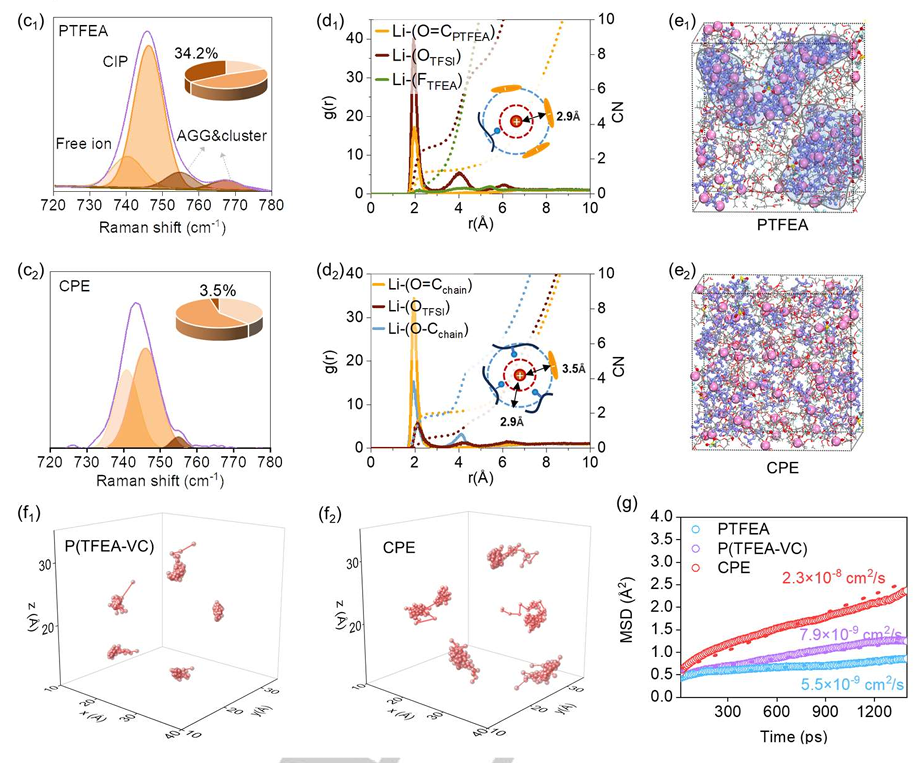
在本研究中,MD计算对理解三元共聚物固态电解质(CPE)中Li⁺的扩散行为起到了关键作用。研究者通过MD模拟计算了CPE与PTFEA电解质中Li⁺的扩散系数,揭示了侧链设计对离子迁移的影响。
具体来说,图3e1和3e2展示了CPE和PTFEA电解质中Li⁺的迁移轨迹,结果表明在CPE中,Li⁺的迁移范围显著较大,表现出更高的动态活性,表明CPE中的离子扩散比PTFEA电解质更加高效。
进一步通过图3g的均方位移(MSD)曲线对比,CPE中的Li⁺扩散系数为2.3 × 10⁻⁸ cm²/s,显著高于PTFEA的5.5 × 10⁻⁹ cm²/s和P(TFEA-VC)的7.9 × 10⁻⁹ cm²/s,验证了CPE在离子导电性方面的显著优势。
这些结果表明,PVC和PEGMEA侧链的引入通过改善离子解离和促进链段运动,显著提高了Li⁺的扩散速率,从而提升了电解质的整体离子导电性。通过MD计算,研究人员能够定量分析并优化固态电解质中的离子扩散,进一步指导高性能电池材料的设计。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!