

机器学习势函数是一种基于数据驱动的势能面近似方法,旨在替代传统经验势函数或量子力学计算。
其核心思想是通过机器学习模型(如神经网络、核方法等)将原子构型映射为系统的总能量和原子受力。
与传统力场不同,MLPs不依赖于预设的物理公式,而是直接从高精度量子力学计算(如密度泛函理论,DFT)生成的数据中学习势能面的复杂特征。
例如,神经网络势函数通过多层非线性变换,将原子环境描述符(如对称函数)与局域原子能量关联,最终通过原子能量求和得到系统总能量。
这种方法的优势在于能够捕捉多体相互作用和非线性效应,适用于复杂材料系统和动态过程模拟。

Chem. Rev. 2021, 121, 16, 10142–10186https://doi.org/10.1021/acs.chemrev.0c01111
机器学习势函数的构建方法
构建机器学习势函数通常包含以下关键步骤:
1.数据准备与特征工程
训练数据通常来自DFT计算,包含不同原子构型对应的能量、力和应力等信息。
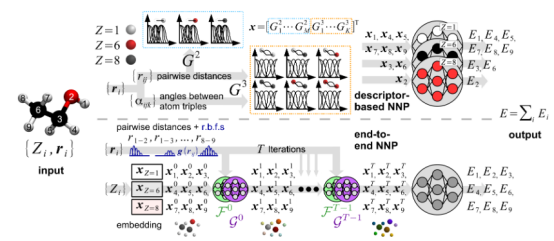
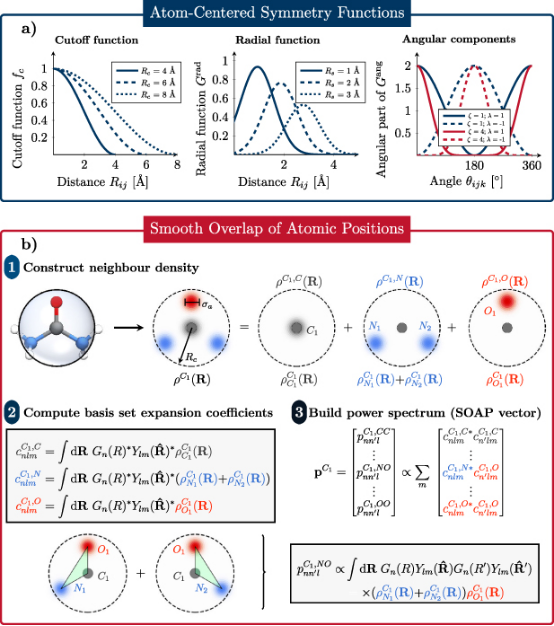
为了将原子环境转化为机器学习模型可处理的输入,需设计满足平移、旋转和置换不变性的描述符。
例如,对称函数(Symmetry Functions)通过径向和角度分布函数编码原子周围的局部环境,而SOAP(Smooth Overlap of Atomic Positions)描述符则通过原子密度投影的功率谱实现更精细的环境表征。
这些描述符需在保留物理意义的同时,降低数据维度以避免过拟合。
J. Phys.: Condens. Matter2025 37 073002,DOI 10.1088/1361-648X/ad9657
2.模型选择与训练策略
常用的模型包括神经网络、高斯过程回归和线性基函数展开等。
例如,Xie等人提出了一种基于正则化线性回归和立方B-样条基的势函数,通过有效分离二体与三体相互作用项,显著提升计算速度。
训练时需平衡能量与力的预测误差,常采用混合损失函数(如均方误差与平均绝对误差的组合)以兼顾异常值鲁棒性和整体精度。
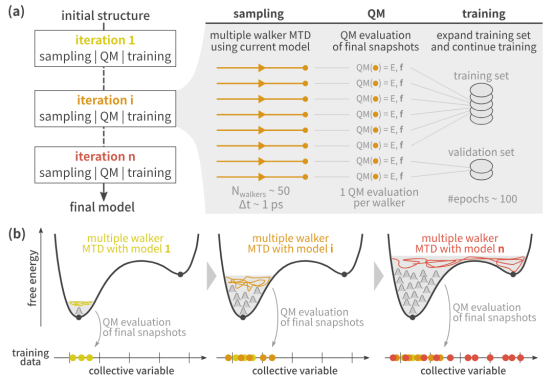
此外,增量学习策略被用于金属有机框架(MOFs)等复杂系统,通过迭代采样与在线训练逐步扩展势函数的适用范围。
npj Computational Materials 2023 9:19 ; https://doi.org/10.1038/s41524-023-00969-x
3.验证与优化
模型性能需通过测试集的能量/力误差、以及衍生性质(如声子谱、弹性常数)与实验或DFT结果的一致性来评估。
例如,某些研究采用主动学习(Active Learning)动态补充训练数据,重点关注势能面中未充分覆盖的区域,以提高模型的泛化能力。
npj Comput Mater9, 162 (2023). https://doi.org/10.1038/s41524-023-01092-7
机器学习势函数的可计算性质
MLPs能够复现或预测多种物理化学性质,涵盖静态与动态特性:
1.结构性质
在材料科学的研究领域中,晶格常数、弹性常数以及表面能等结构性质,是理解材料微观结构和宏观性能的关键参数。
这些参数可以通过能量对原子位置的二阶导数进行精确计算。
例如,在对钨这种金属材料的模拟过程中,机器学习势函数(MLPs)展现出了强大的预测能力。
它不仅成功地预测了负空位结合能,还能够详细地描述位错结构的细节,为深入研究钨的力学性能和微观缺陷提供了有力的支持。

Phys. Rev. Materials 5, 103803 DOI: https://doi.org/10.1103/PhysRevMaterials.5.103803
2.热力学性质
热力学性质对于研究材料在不同条件下的行为至关重要,其中熔点和相变行为是核心内容。
这些性质可通过分子动力学模拟获得,模拟过程中能够动态地观察材料内部原子的运动和相互作用。
如 Zong 等人(2020)开展的研究工作,他们利用分层的机器学习势函数(MLPs)对高压氢进行了深入研究,成功揭示了高压氢的液态密度反常现象。
这一发现不仅加深了我们对氢在极端条件下物理性质的理解,也为相关领域的研究提供了新的思路和方法。
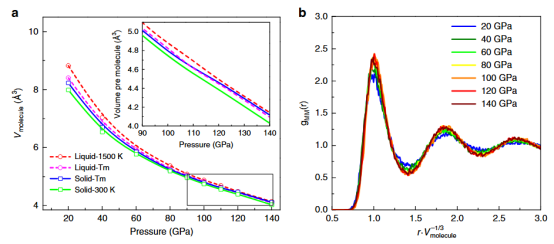
Nat Commun11, 5014 (2020). https://doi.org/10.1038/s41467-020-18788-9
3.动力学性质
扩散系数、粘度、声子谱等动力学性质与原子的运动密切相关,能够反映材料内部的动态过程。
这些特性可通过长时间分子动力学模拟提取,在模拟中跟踪原子的运动轨迹并分析其统计规律。
Benoit 等人(2019)所构建的机器学习势函数(MLPs)在这方面表现出色,它准确地复现了金铁合金的声子色散曲线。
这一成果对于理解金铁合金的热学、电学等性能具有重要意义,也验证了 MLPs 在研究动力学性质方面的有效性和可靠性。
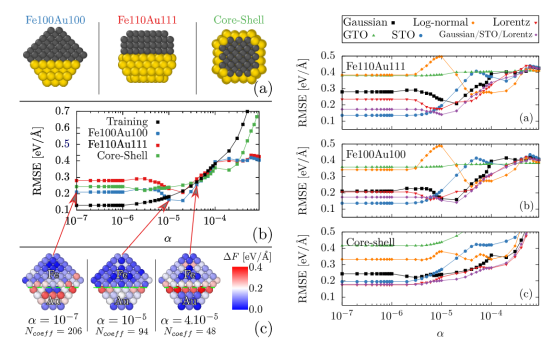
Mach. Learn.: Sci. Technol.2 025003DOI 10.1088/2632-2153/abc9fd
4.化学反应与缺陷行为
机器学习势函数(MLPs)在模拟化学反应与缺陷行为方面具有独特的优势。
它可以模拟点缺陷形成能、化学反应路径及电荷转移过程等重要信息。
例如,Karim 等人(2023)开发了一种统一势函数,该函数能够自主捕捉 SiO₂非晶相中的缺陷重构行为。
通过对缺陷重构行为的模拟和分析,可以更好地理解 SiO₂材料的微观结构演变和性能变化,为优化材料性能和设计新材料提供理论依据。
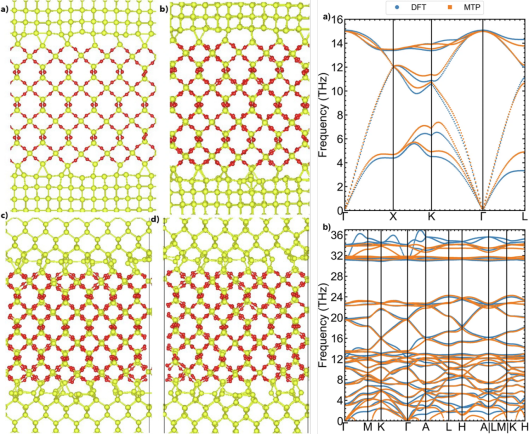
npj Comput Mater10, 218 (2024). https://doi.org/10.1038/s41524-024-01390-8
5.自由能与化学势
自由能和化学势是描述物质状态和反应方向的重要热力学参数。
结合热力学积分方法,机器学习势函数(MLPs)可计算溶剂化自由能和真实化学势。
如 Jinnouchi(2024)通过机器学习辅助方法,精确预测了水溶液中离子的化学势。
这一成果不仅为研究水溶液中的化学反应和离子行为提供了准确的热力学数据,也拓展了 MLPs 在热力学领域的应用范围,为解决复杂体系的热力学问题提供了新的途径。
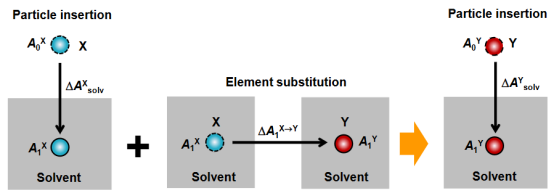
J. Chem. Phys. 161, 194110 2024https://doi.org/10.1063/5.0240275
文献案例分析:超快速可解释机器学习势函数
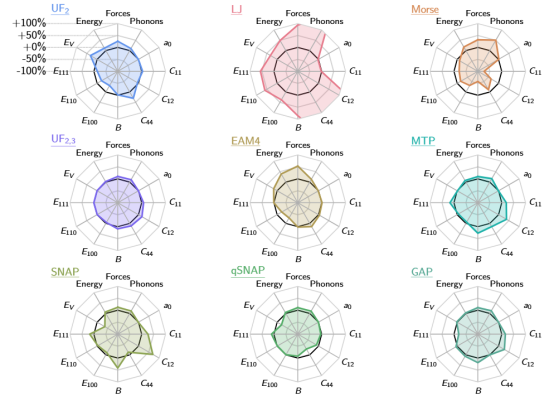
以Xie等人(2023)的《Ultra-fast interpretable machine-learning potentials》(npj Comput Mater9, 162 (2023). https://doi.org/10.1038/s41524-023-01092-7)为例,该研究提出了一种基于线性回归与B-样条基的高效MLP框架,主要贡献包括:
1.方法创新
通过将势函数分解为二体与三体相互作用项,并采用正则化线性回归拟合立方B-样条系数,模型在保证可解释性的同时,计算速度比主流MLPs快2-4个数量级。
这种设计显著降低了优化问题的复杂度,使训练时间从数天缩短至数小时。
2.性能验证
在钨和硅等材料的测试中,该势函数预测的能量与力误差与SNAP、MTP等先进模型相当,而计算耗时仅略高于传统经验势。
例如,对于DFT数据集,其预测的弹性常数误差小于5%,声子谱与实验数据高度吻合。
此外,该模型在稀疏训练集下仍保持良好精度,展示了优异的泛化能力。
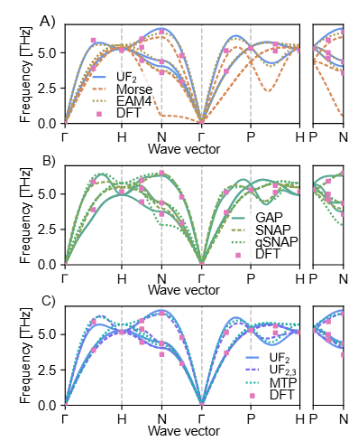
3.应用拓展
该势函数被用于模拟钨的熔点和铁碳合金的缺陷动力学,结果与实验观测一致。
作者进一步指出,通过引入四体相互作用项,可扩展至有机分子体系,为复杂系统的多尺度模拟提供了新思路。
结论
机器学习势函数通过融合数据驱动方法与物理约束,正在重塑原子尺度模拟的范式。
其在计算效率与精度间的平衡能力,使其成为研究材料相变、缺陷动力学和化学反应的有力工具。
然而,模型的可解释性、对极端条件的泛化能力以及多组分系统的适用性仍需进一步探索。
未来,随着主动学习与高吞吐量计算的结合,MLPs有望在更广阔的化学空间和更长的时间尺度上实现突破。