

说明:在分子动力学、蒙特卡洛等计算模拟领域,势函数作为描述原子/ 分子间相互作用的数学模型,犹如连接微观原子行为与宏观材料性能的 “物理引擎”,其选择直接决定模拟的精度、效率与物理真实性。
然而,面对金属、半导体、聚合物等多元材料体系,以及从静态结构优化到动态化学反应的复杂需求,研究者常面临 “模型陷阱”—— 经典势函数可能因忽略多体效应而失真,高精度计算又受限于算力瓶颈。本文将从势函数的分类逻辑、选择框架、实战案例及前沿趋势展开,为复杂体系的模拟建模提供系统化解决方案。

势函数的多维分类:从经验简化到数据智能
经典势函数基于物理经验构建,通过简化假设实现计算效率与模型精度的平衡。对势如Lennard-Jones 适用于惰性气体或简单金属(如 Ar、Cu),仅描述两体相互作用,虽能快速模拟液态金属相变,却无法捕捉金属键的离域特性;多体势如嵌入原子势(EAM)则通过 “电子密度嵌入能” 刻画金属键的协同效应,被广泛用于铝、铁等金属的塑性变形模拟,可准确计算层错能、表面能等关键参数,但参数化高度依赖实验或 DFT 数据。
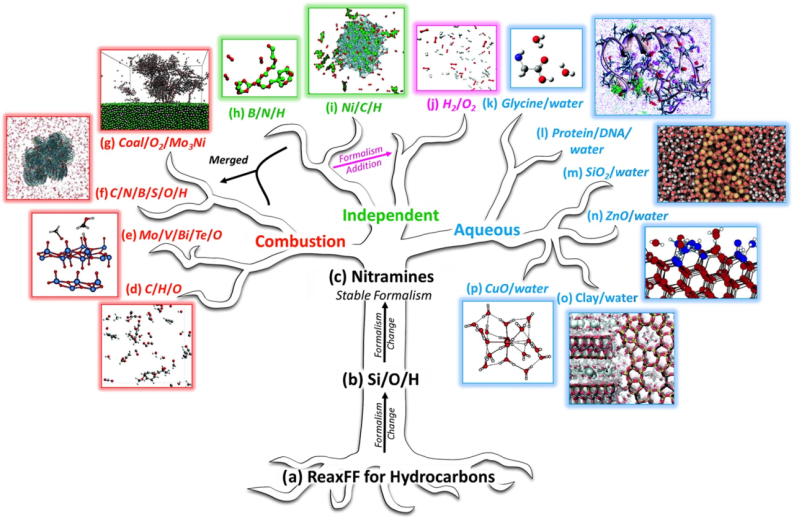
半经验势与反应力场则迈向化学真实性,例如 ReaxFF 通过动态键级更新模拟化学键断裂与形成,适用于燃烧、催化等反应体系,而 COMB 势通过电荷自洽优化提升氧化物体系的模拟精度。
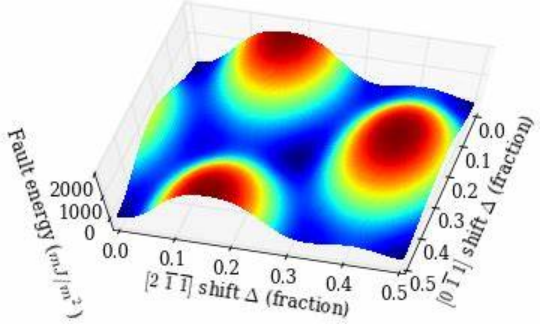
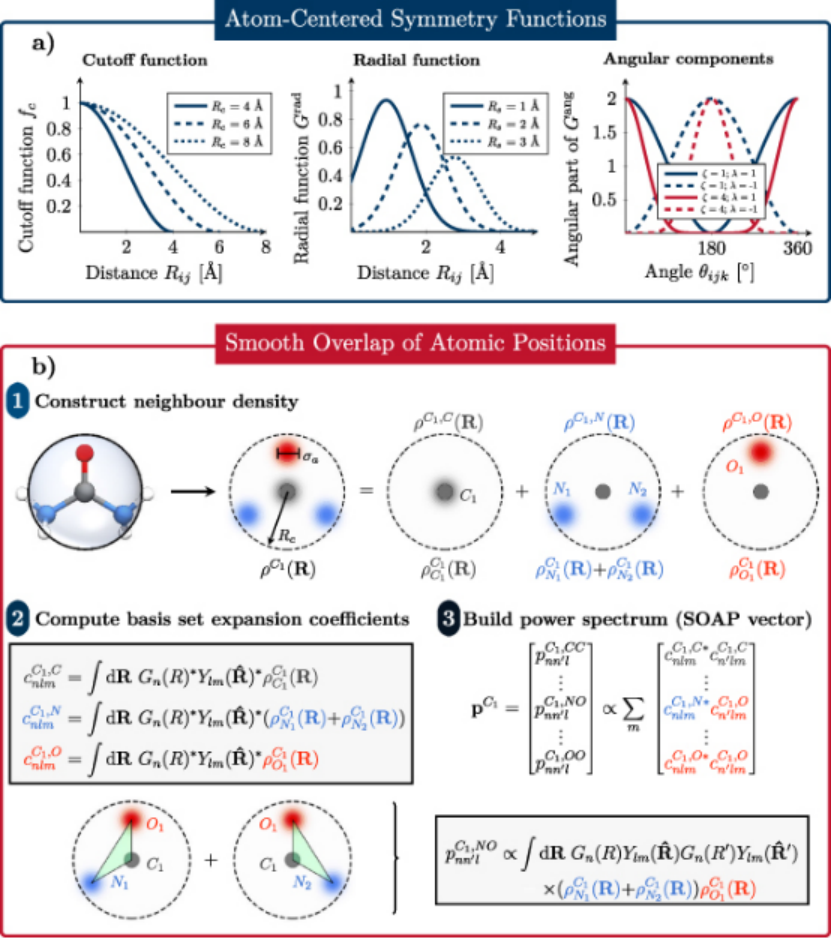
机器学习势函数标志着模拟进入数据驱动时代。以DeepMD、ANI 为代表的神经网络势通过深度神经网络拟合 DFT 数据,可精准还原复杂体系的势能面,如训练后的硅势函数能同时再现晶体硅的弹性模量与非晶硅的扩散系数,但需数十万原子级构型作为训练数据,且实时模拟依赖 GPU 加速。
高斯过程势(GAP)则在小数据集下更具优势,通过贝叶斯推断量化预测不确定性,适合非晶态材料或新型合金的快速建模。第一性原理势如 DFT 虽为 “黄金标准”,却受限于计算规模(通常≤1000 原子),更多作为基准工具验证其他模型的可靠性。
DOI10.1088/1361-648X/ad9657
选择框架:四维评估体系的构建
选择势函数需综合材料特性、计算目标、数据支撑与硬件资源。在材料体系维度,金属 / 合金优先考虑 EAM 或机器学习势 —— 如模拟铝单晶位错运动时,Zhou-Al EAM 势因准确描述层错能(与实验值偏差<5%)成为首选;共价材料如 Si、C 体系适用 Tersoff 势,而涉及化学反应的半导体界面(如 Si/SiO₂热输运)则需混合 Tersoff 与 ReaxFF 势,通过 LAMMPS 的 “hybrid” 命令实现跨尺度兼容。
https://doi.org/10.1038/npjcompumats.2015.11
精度与效率的权衡贯穿选择全程:快速筛选阶段可采用经典势或紧束缚模型(如TB)在微秒级尺度扫描合金相图;关键界面或反应路径则需 DFT 或神经网络势校准 —— 例如异质结肖特基势垒计算中,先通过 EAM 势定位缺陷敏感区域,再用 DFT 精确计算电荷密度差。
参数化与验证是模型可信性的基石,需优先调用 OpenKIM、NIST 等开源数据库的认证势函数,并通过晶格常数、扩散系数等静态与动态指标对比实验或 DFT 数据,如 Si 的晶格常数误差需控制在 1% 以内。
软件兼容性与计算资源同样关键。LAMMPS 对经典势支持全面,适合大规模金属塑性模拟;GROMACS 内置生物分子力场(如 CHARMM),更适合聚合物体系;机器学习势的部署则需深度集成框架(如 DeepMD-kit 与 LAMMPS 对接),并配置 GPU 加速环境以应对高计算负载。
实战场景:从原子模拟到器件建模的策略
在金属塑性变形模拟中,EAM 势展现出不可替代的优势。以铝单晶拉伸为例,Zhou-Al EAM 势通过准确描述原子间多体相互作用,模拟得到的层错能(0.04 eV/Ų)与实验值(0.038 eV/Ų)高度吻合,进而可靠预测位错运动速度与孪晶形成机制,为铝合金强度优化提供理论依据。
https://doi.org/10.1016/j.commatsci.2019.109432
半导体界面热输运模拟则需混合势的协同作用。在 Si/SiO₂体系中,Si 区域采用 Tersoff 势刻画共价键,SiO₂区域通过 ReaxFF 势模拟离子键,界面处通过势函数混合技术实现力场兼容。
通过对比 DFT 计算的声子散射谱,调整键长参数以匹配电荷分布,最终模拟得到的界面热导率与实验值偏差<8%,有效揭示粗糙度对声子散射的抑制效应。
高分子材料的跨尺度模拟凸显粗粒化策略的价值。在聚乙烯链拉伸断裂研究中,小尺度(<1000 原子)采用 AIREBO 势捕捉 C-C 键断裂的化学细节,而大尺度(>10⁵原子)切换至 MARTINI 粗粒化势,将每个链节映射为珠子,模拟速度提升千倍的同时,仍能再现应力 – 应变曲线的屈服平台特征。
电催化反应模拟则是机器学习势的突破领域。传统经典势无法描述 Pt 表面 CO 吸附诱导的重构效应,而预训练的 Pt-O-C 神经网络势(如 DeepPtOX 模型)可精准再现 O₂解离能垒(0.7 eV vs. 实验 0.68 eV),并通过分子动力学模拟揭示同位素效应下的反应路径差异,为高效催化剂设计提供原子级洞察。
https://doi.org/10.1016/j.surfin.2025.106542
挑战应对与未来图景
势函数在极端条件(如高温、高压)下的失效问题,本质是参数外推局限性的体现。例如金属熔沸点模拟偏差超20% 时,需切换至高温校准的机器学习势或引入温度依赖的键级参数(如 ReaxFF)。
多元素体系参数缺失时,可通过 OpenKIM 的 “势函数嫁接” 技术组合二元势,或利用遗传算法自动优化三元势参数,以 DFT 计算的合金形成焓为目标函数迭代修正。
计算效率瓶颈的突破依赖多技术融合:主动学习策略通过少量DFT 数据迭代更新神经网络势,降低训练成本;“冷热区” 多尺度方法将反应活跃区域用 NNP 精细模拟,惰性区域用粗粒化势简化,实现计算资源的智能分配。
https://doi.org/10.1016/j.fuel.2024.133650
未来,势函数发展将呈现三大趋势:自动化生成工具(如DeePMD-kit)通过贝叶斯优化实现 “数据输入 – 模型输出” 的全流程无人化;量子 – 经典 – 机器学习的三级耦合框架(DFT – 神经网络势 – 粗粒化模型)将打通从电子结构到宏观性能的跨尺度模拟;标准化开源生态(如 OpenKIM 认证体系)则通过格式统一与社区协作,推动势函数从 “定制化开发” 走向 “即插即用”。
结语:在简化与真实间寻找平衡
势函数的选择是一场“带着枷锁的舞蹈”—— 既要用有限的数学模型逼近无限的物理真实,又需在计算资源与时间成本间寻找最优解。
研究者需以材料化学直觉为罗盘(如键型分析、极性判断),以数据验证为锚点(如晶格常数、反应能垒),构建 “假设 – 模拟 – 验证 – 修正” 的闭环思维。
随着机器学习与高通量计算的赋能,势函数正从 “人工设计的艺术品” 进化为 “数据驱动的工业品”,未来的材料研发或将迎来 “输入元素列表 – 智能生成势函数 – 精准预测性能” 的范式革命,让模拟真正成为连接理论设计与实验制备的无缝桥梁。