

机器学习势函数(Machine Learning Potentials, MLPs)是一种基于数据驱动的势能面近似方法,旨在替代传统经验势函数或量子力学计算,以更高效、更准确地模拟原子和分子系统的物理行为。其核心思想是通过机器学习算法(如神经网络、高斯过程回归等)将原子构型映射为系统的总能量和原子受力,从而构建一个能够高效预测原子间相互作用势的模型。
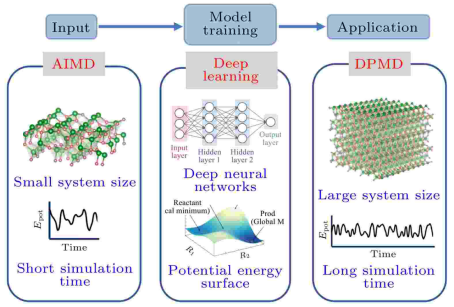

基本原理
机器学习势函数的基本原理是通过训练数据集中的第一性原理计算(如密度泛函理论DFT)能量和力,构建一个能够近似势能面(Potential Energy Surface, PES)的模型。这种模型可以高效地预测材料的性质,如弹性常数、相变过程等,并且在某些情况下,其预测精度可与第一性原理计算相媲美。
机器学习势函数的构建通常包括以下几个步骤:
数据收集:收集第一性原理计算得到的原子构型、能量和力的数据。
描述符选择:选择合适的描述符(Descriptors)来描述原子的化学环境。常用的描述符包括原子坐标、邻近原子类型、距离等。
模型训练:使用机器学习算法(如神经网络、高斯过程回归等)训练模型,使其能够根据输入的原子构型预测系统的总能量和原子受力。
模型验证:通过测试集的能量/力误差、以及衍生性质(如声子谱、弹性常数)与实验或DFT结果的一致性来评估模型性能。
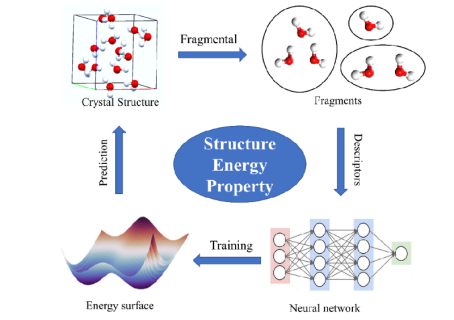
模型选择与训练策略
常用的机器学习模型包括神经网络、高斯过程回归和线性基函数展开等。例如,Xie等人提出了一种基于正则化线性回归和立方B-样条基的势函数,通过有效分离二体与三体相互作用项,显著提升计算速度。
训练时需平衡能量与力的预测误差,常采用混合损失函数(如均方误差与平均绝对误差的组合)以兼顾异常值鲁棒性和整体精度。此外,增量学习策略被用于金属有机框架(MOFs)等复杂系统,通过迭代采样与在线训练逐步扩展势函数的适用范围。
化学反应与缺陷行为
机器学习势函数(MLPs)在模拟化学反应与缺陷行为方面具有独特的优势。它可以模拟点缺陷形成能、化学反应路径及电荷转移过程等重要信息。例如,SchNet是一种专门为分子和材料设计的深度学习架构,通过连续滤波卷积层来建模原子间的相互作用。SchNet能够准确预测一系列化学性质,并且在分子动力学模拟中表现出色。
应用领域
机器学习势函数在多个领域得到了广泛应用,包括:
材料科学:用于模拟材料的力学性能、热输运特性等。例如,山东高等技术研究院郭瑞强研究员课题组利用机器学习势函数研究了六方氮化硼中四玻色散射对各向异性热输运的影响。
化学反应:用于模拟化学反应路径及电荷转移过程。例如,MedeA MLP(机器学习势函数)是基于LAMMPS的机器学习势函数模拟的完整MedeA支持,它利用机器学习方法挖掘和利用第一性原理数据集,以插值和推测的方式提高模拟效率和准确性。
与其他方法的对比
机器学习势函数与传统经验势函数和第一性原理计算相比,具有以下优势:
计算效率:相比第一性原理计算,机器学习势函数的计算成本显著降低,但仍能保持较高的精度。
适用性:机器学习势函数对元素种类限制较少,更适合多元素体系的模拟。
可扩展性:通过增量学习策略,机器学习势函数可以逐步扩展其适用范围,适用于更复杂的系统。
未来展望
随着计算机算力和人工智能技术的迅猛发展,机器学习势函数在材料科学、化学反应模拟等领域展现出广阔的应用前景。例如,深度势能方法(Deep Potential)是一种典型的机器学习势函数方法,能够构建同时满足第一性原理精度和经验势函数效率的模型,解决传统方法难以解决的问题。此外,生成模型(如扩散模型)也被用于探索晶体材料空间,为未来的研究提供了新的思路。
总结
机器学习势函数是一种基于数据驱动的势能面近似方法,通过结合机器学习算法与量子力学计算数据,构建能够高效预测原子间相互作用势的模型。它在材料科学、化学反应模拟等领域具有广泛的应用前景,并且在计算效率和精度方面表现出显著优势。随着技术的不断发展,机器学习势函数将在更多领域发挥重要作用。