Gaussian 凭借其在孤立分子与小团簇建模中的高精度与高效率,适用于反应路径构建、过渡态搜索与轨道电子结构解析,尤其适合有机反应、均相催化与金属配合物体系。
VASP 则在周期性边界条件建模、晶体表面吸附行为模拟及电子态密度分析方面具有显著优势,是异相催化、电催化与材料电子调控研究的主力平台。
文章通过具体文献案例说明两者在反应路径、自由能分析及催化活性预测中的典型应用场景,为科研人员依据研究对象与目标选择合适建模工具提供理论参考与实践指导。
密度泛函理论(Density Functional Theory,DFT)已成为当今催化科学研究中不可或缺的理论工具。作为一种从量子力学基本原理出发构建的电子结构计算方法,DFT 不仅能够解释催化反应过程中的热力学和动力学现象,还能预测新材料、新催化剂的潜在性能,从而有效辅助实验设计与筛选。
随着 DFT 方法与高性能计算资源的不断发展,其在催化反应路径解析、吸附行为预测、电子结构调控等方面的作用愈发突出。
在催化研究的实际操作中,Gaussian 和 VASP 是使用最为广泛的两种 DFT 计算软件。二者均已发展成熟,理论框架完备,但它们的计算方法和应用对象有着显著的差异。
Gaussian 采用原子轨道展开方法,适用于分子体系与非周期结构的高精度量子化学计算;VASP 基于平面波方法和周期边界条件,适合模拟晶体材料、金属表面及其它周期性体系中的电子结构行为。
在具体研究中,如何根据体系类型与研究目标,合理选择 Gaussian 或 VASP,成为理论建模中最关键的环节之一。不少研究者在项目实施过程中往往遇到这样的问题:“反应路径是否该用 Gaussian 来计算?材料表面是否必须使用 VASP?”“同一个反应,能否用两个工具协同建模?”“如何分配计算任务,既高效又准确?”
这些看似技术性的问题,其实根本指向的是对研究对象本质、反应机制与材料效应理解的深入与否。
本文将围绕上述问题,从应用体系、建模任务与功能侧重点三个层面系统梳理 Gaussian 与 VASP 的异同及适用边界。我们将着重分析它们在催化领域中分别适合解决什么样的科学问题,并从实际操作角度给出研究设计建议,帮助科研人员构建更高效、更精准的催化反应建模方案。
Gaussian 是当前量子化学领域使用最为广泛的软件之一,其核心优势在于对孤立分子、分子团簇、小金属配合物等非周期体系的高度适应性。
Gaussian 所采用的原子轨道展开法使其在结构优化、能量计算与反应路径分析方面具有非常高的计算效率和可靠性,因此广泛应用于均相催化、金属团簇催化、有机反应机制等研究场景。
Gaussian 在处理非周期体系结构优化方面具有很强的稳定性和效率。其可通过基态优化获得反应物、中间体与产物的最优几何构型,为进一步的路径分析与过渡态搜索提供必要前提。
借助其频率分析功能,研究者不仅可以判断结构是否为极小值(无虚频)或过渡态(存在一个虚频),还可以获取 Gibbs 自由能、焓、熵等热力学参数,进而评估反应在特定温度下的可行性和热力学优势。
在催化研究中,这一功能尤为关键。例如,在研究 CO₂ 还原、氨合成等典型反应过程中,不同中间体的自由能变化可用于筛选优先路径,热力学稳定性也为实验选择合适反应条件提供理论参考。
Gaussian 强大的过渡态搜索功能是其区别于多数 DFT 软件的另一大亮点。其支持使用 QST2、QST3、STQN 等算法,在已知反应物与产物的基础上定位过渡态结构。配合 IRC(Intrinsic Reaction Coordinate)路径跟踪分析,Gaussian 可用于确认反应路径是否真实、连续,确保过渡态确实连接相应的前后构型。
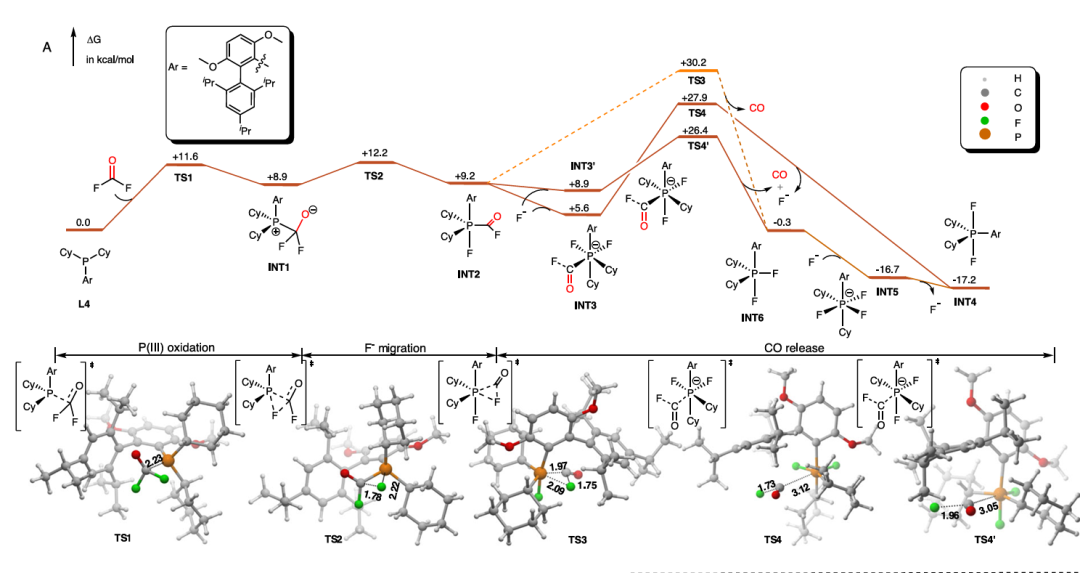
在反应机制探索中,过渡态结构是判断速率控制步骤与活性位点的核心信息。例如,在 Zhao 等人(2023)发表于 Nature Communications 的研究中,作者开发了一种 Pd(II)/Pd(0) 与 P(III)/P(V) 协同催化的氟羰基化反应,成功将三氟硼酸钾类底物高效转化为酰氟。
该研究的机理验证依赖于 Gaussian 软件进行的 DFT 计算,不仅验证了 Pd(II)-OCF₃ 中间体的稳定性,还计算出 β-F 消除路径的活化能(ΔG‡ = 8.4 kcal/mol),远低于直接还原消除路径(ΔG‡ = 40.5 kcal/mol),从而解释了实验中酰氟产物的专一性生成。
此外,该研究还通过计算揭示了磷配体(如 Brettphos)与 COF₂ 反应生成 CO 的详细机制,阐明了配体在反应中“双重角色”的电子转移路径。这种基于 Gaussian 的 DFT 支持,不仅帮助确认了反应路径的可行性与中间体的真实性,也通过过渡态构型与势能面的构建,为实验方案提供了可靠的理论支持。
Gaussian 提供的过渡态优化及路径追踪功能,大幅增强了反应路径建模的可信度,特别适用于气相反应、均相催化体系以及金属团簇催化过程的初步筛选与构型确认。
除了热力学与反应路径建模外,Gaussian 在分子电子结构分析方面也有显著优势。通过前线轨道(HOMO/LUMO)、自然布居分析(NBO)、Mulliken 电荷分析等工具,研究者可以定量分析电子密度分布、反应活性中心、电荷转移路径及成键特性。
这些结果对于解释催化选择性、催化剂调控机制及反应物电子匹配度具有重要意义。特别是在研究金属配合物催化剂时,不同配体的电子给/取能力对反应路径与能垒的调节可通过 Gaussian 的轨道分析精确刻画,从而为配体设计提供理论依据。
对于具有催化活性的金属团簇(如 M₁₃、Pt₂₀ 等),Gaussian 提供了一个高效建模与计算的解决方案。在不依赖周期性边界条件的情况下,Gaussian 可对团簇与反应物的相互作用进行吸附能计算、几何优化与反应路径扫描。
这种方式特别适用于研究团簇催化剂对 CO₂、N₂、O₂ 等小分子的活化机制,或用于快速筛选不同团簇尺寸与构型的活性差异。
相较于 Gaussian,VASP 的理论基础和应用重心完全不同。VASP 采用平面波基组与周期性边界条件,能够自然建模固体晶体、金属表面、二维材料等周期性材料体系,是固态催化与异相反应研究的主力工具。
在催化剂材料研究中,表面结构对反应行为有着决定性作用。VASP 可构建周期性 slab 模型以模拟实际表面环境,通过截取晶胞、添加真空层、设置层间约束等方式,构建真实金属表面、氧化物载体或界面结构。随后,采用结构优化方法释放表面原子构型,从而得到稳定的表面结构,作为吸附计算与反应路径建模的起点。
这一建模过程可以灵活引入表面缺陷、掺杂原子、界面结构等,拓展研究对象的复杂性与现实性,为研究真实催化剂的工作机制提供可计算模型。
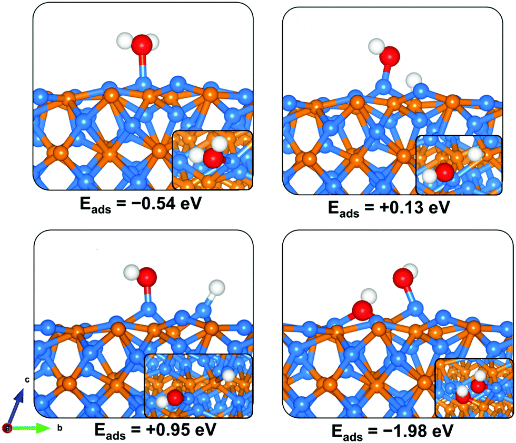
在实际催化反应中,吸附行为是反应启动的第一步。VASP 允许在表面多个位点上放置反应物分子,通过结构优化获取吸附构型,进而计算吸附能(E_ads = E_slab+molecule – E_slab – E_molecule)。
不同构型间吸附能的差异可揭示反应物优选吸附位点、可能形成的中间体结构等,有助于分析催化剂的选择性与催化活性来源。
吸附构型也是后续进行 CI-NEB 反应路径计算的重要前置条件。在电催化和表面催化研究中,吸附能还常被用于构建微观动力学模型,如 Sabatier 分析图谱。
在非均相催化中,VASP 通常承担两个关键任务:一是计算表面反应的反应能垒与路径,二是绘制自由能台阶图以评估热力学可行性和选择性。
首先,VASP 通过 Climbing Image-NEB (CI-NEB) 方法对表面反应路径进行建模。在该方法中,研究者预设反应物与产物构型,并在路径中插入多个“图像结构”,在优化过程中自动逼近反应坐标空间的最小能量路径,进而获得过渡态及反应能垒。
该方法适用于如 *CO → *CHO、*O → *OH 等典型的表面反应步骤,是研究表面动力学机制不可或缺的工具。
同时,VASP 更广泛的用途是绘制 自由能台阶图(Free Energy Diagram)。研究者通过优化不同吸附态的构型,计算其吸附能,并结合零点能、熵项与电极电势修正,最终得到 Gibbs 自由能变化(ΔG)。
这些数据被串联构成反应路径上每一步的自由能变化,从而识别出速率控制步骤(RDS)与有利的反应路径。这种方法在 HER、OER、CO₂RR 等研究中尤为常见,也是材料筛选与活性比较的理论基础。
相较于 Gaussian 多用于小分子体系的 ΔG 计算,VASP 的自由能分析更强调吸附态、表面结构和电极条件的耦合影响,在表面催化和电催化领域具有更强的实际适用性。
在周期性体系中,材料的电子结构直接决定其催化性能。VASP 可计算总态密度(DOS)、投影态密度(PDOS)、电荷密度分布、功函数、d 带中心位置等电子结构特征,这些参数被广泛用于解释吸附行为、调控催化活性、识别活性中心等。
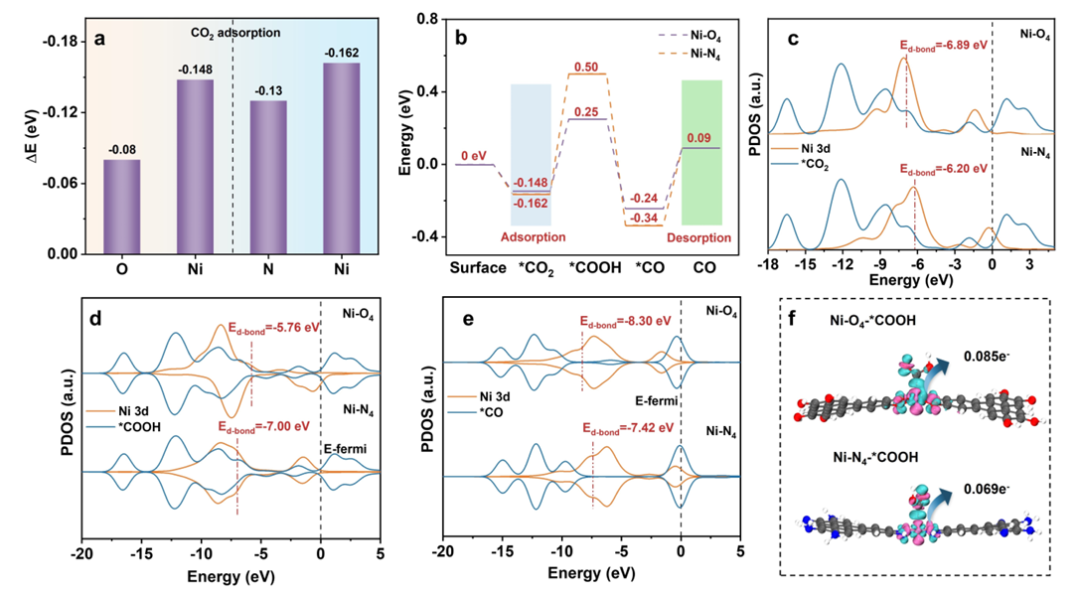
以 Liu 等人(2024)在 Angewandte Chemie 上的研究为例,作者基于导电金属有机框架(CMOFs)设计了两种具有不同配位环境的单原子 Ni 位点(Ni-O₄ 与 Ni-N₄),并通过 VASP 模拟揭示其在 CO₂ 光催化还原中的活性差异。
研究显示,Ni-O₄ 中 Ni 位点的 d 带中心显著上移,接近费米能级,使其对关键中间体 *COOH 的结合更稳定、形成势垒更低(ΔG = 0.398 eV),同时也有利于 *CO 的快速脱附(ΔG = 0.33 eV)。
该电子结构调控策略,不仅实现了对中间体转化路径的优化,也赋予 Ni-O₄ CMOFs 在稀释 CO₂(例如烟道气)环境下更强的选择性与稳定性。
此外,VASP 构建的态密度与差分电荷分布图进一步表明,Ni-O₄ 的配位结构诱导出更强的 p-d 杂化效应,Ni 3d 轨道与 O 2p 轨道间耦合增强,从而增强了其对 *COOH 的电子耦合能力;而对产物 CO 的吸附则因反键轨道填充度较低而减弱,这正是高选择性与反应速率得以统一的电子本质。
整套工作充分体现出 VASP 在探索“配位工程调控金属中心电子结构,从而实现高效 CO₂ 转化”机制中的不可替代作用。
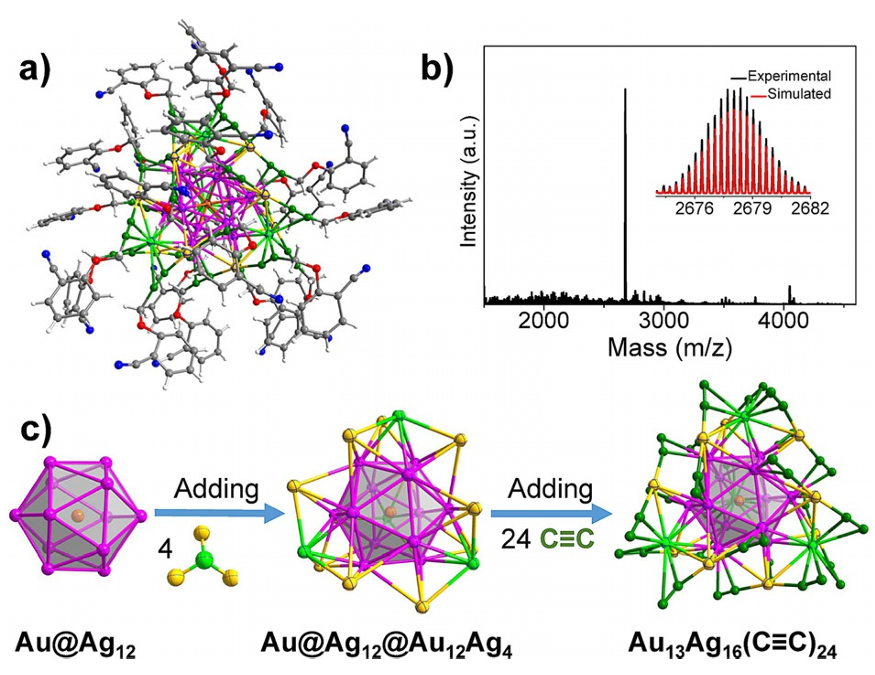
1. Zhaoxian Qin, Sachil Sharma, Chong-qing Wan, Sami Malola, Wen-wu Xu, Hannu Häkkinen & Gao Li. A Homoleptic Alkynyl-Ligated [Au
₁₃Ag₁₆(C₁₀H₆NO)₂₄]³⁻ Cluster as a Catalytically Active Eight-Electron Superatom. Angew. Chem. Int. Ed. 2021, 60, 970–975. DOI:10.1002/anie.202011780
2. Jiahui Liu, Bin Han, Xueming Liu, Shujie Liang, Yang Fu, Jun He, Lai-Hon Chung, Yuanfang Lin, Yupeng Wei, Sibo Wang, Tianyi Ma & Zhifeng Yang. Tailoring d-Band Center of Single-Atom Nickel Sites for Boosted Photocatalytic Reduction of Diluted CO₂ from Flue Gas. Angew. Chem. Int. Ed. 2025, 64, e202417435. DOI:10.1002/anie.202417435
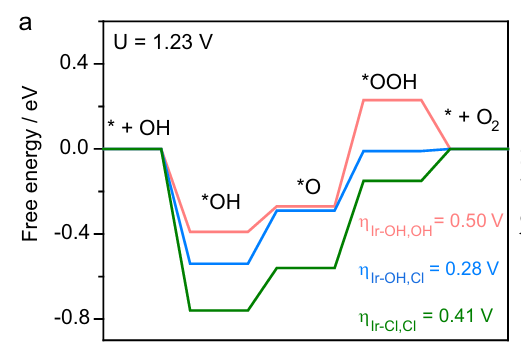
3. Xinxuan Duan, Qihao Sha, Pengsong Li, Tianshui Li, Guotao Yang, Wei Liu, Ende Yu, Daojin Zhou, Jinjie Fang, Wenxing Chen, Yizhen Chen, Lirong Zheng, Jiangwen Liao, Zeyu Wang, Yaping Li, Hongbin Yang, Guoxin Zhang, Zhongbin Zhuang, Sung-Fu Hung, Changfei Jing, Jun Luo, Lu Bai, Juncai Dong, Hai Xiao, Wen Liu, Yun Kuang, Bin Liu & Xiaoming Sun. Dynamic chloride ion adsorption on single iridium atom boosts seawater oxidation catalysis. Nat. Commun. 2024, 15, 1973. DOI:10.1038/s41467-024-46140-y
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!