说明:文章华算科技系统梳理了氢键的定义、分类与核心参数,逐一介绍了XRD、ND、cryo-EM、IR、NMR、Raman等实验手段及DFT、MD、分子对接等氢键计算方法。
氢键不是化学键,而是一种特殊的分子间(或分子内)相互作用,其本质上是一种静电相互作用,是带部分正电荷的氢原子与带部分负电荷的原子之间的吸引力。
氢供体:一个氢原子(H)必须与一个电负性很强、原子半径较小的原子(如 氮N、氧O、氟F)以共价键结合。
氢受体:另一个分子或同一分子的不同部位,必须有一个带有孤对电子的电负性原子(同样是 N, O, F)。
当这两个条件满足时,那个几乎“裸露”的、带部分正电的氢原子核,就会被受体原子上的孤对电子所吸引,从而形成氢键。我们可以将其简写为 X-H···Y,其中X是供体,Y是受体。
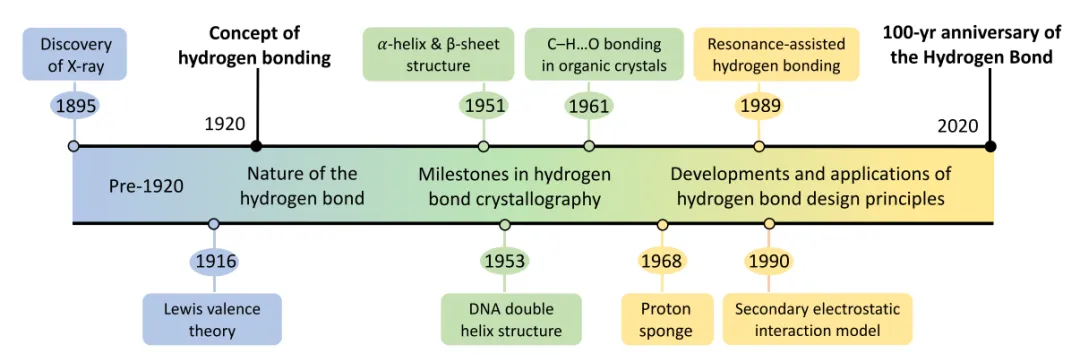
图1. 氢键原理的发展史。DOI:10.1002/wcms.1477
随着研究发现,氢键的供体和受体不必局限于 N、O、F。例如 C-H ,C连有吸电子基团时,H 可带 δ+可作为供体形成 “C-H…O” 氢键,π 键(电子云密集区)可作为受体形成 “X-H…π” 氢键 —— 这些被称为 “非经典氢键”,虽强度较弱,但在蛋白质折叠、有机晶体堆积中同样发挥关键作用。
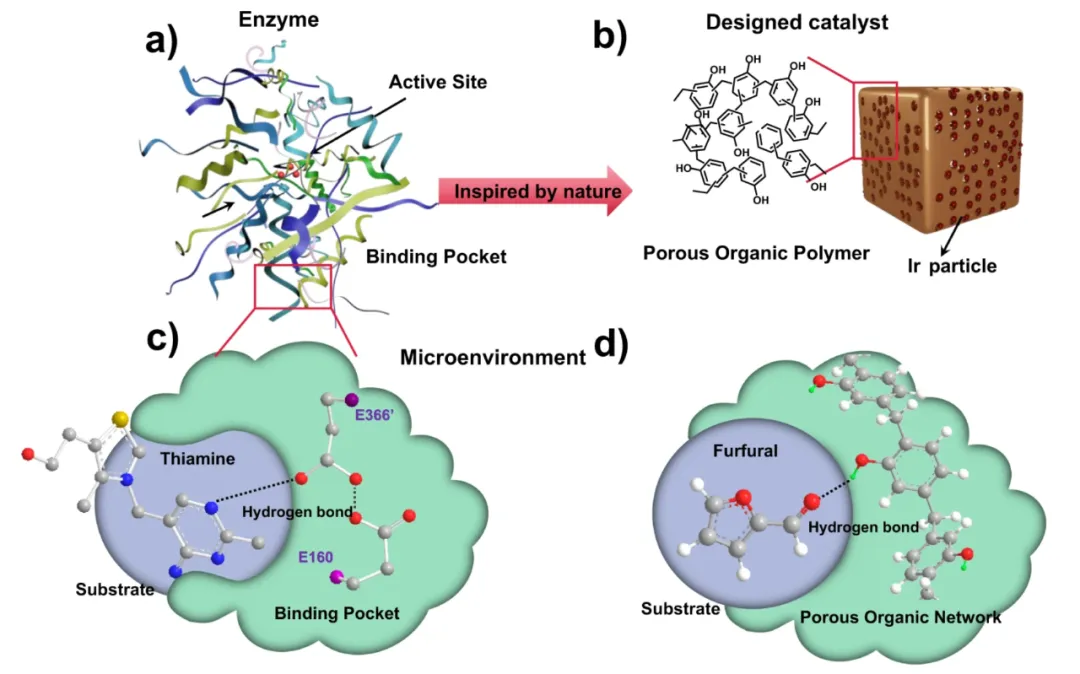
图 2. 氢键网络的作用示意图。DOI:10.1038/s41467-023-36015-z
具有饱和性:一个氢原子只能与一个电负性原子形成一个氢键。因为它只有一个原子核,正电中心只有一个。
具有方向性:氢键的强度与方向有关。当氢供体(H)、氢受体(…O、…N)三者排在一条直线上(键角接近180°)时,静电作用最强,氢键也最稳定。
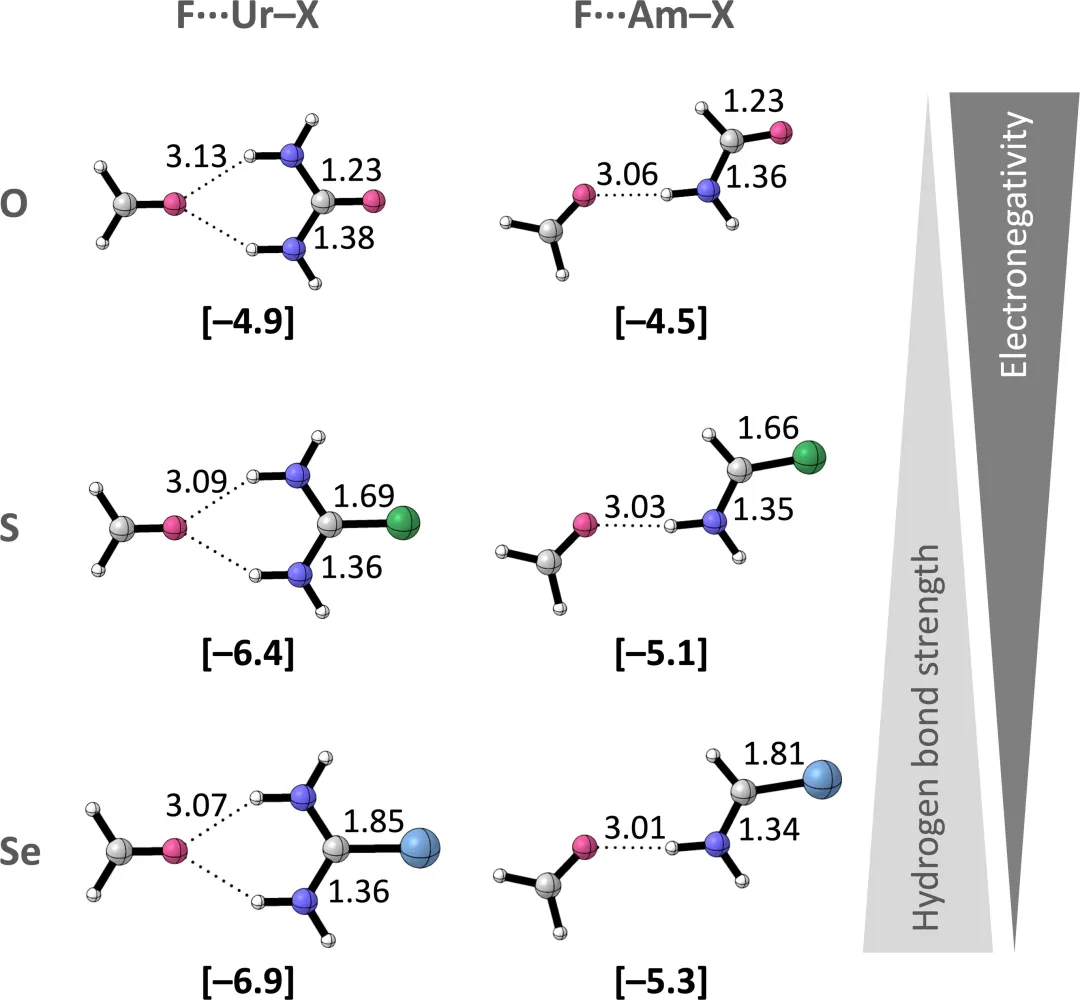
图3. 考虑方向性氢键相互作用的改进的离子液体溶液相行为的预测。DOI: 10.1021/acs.iecr.9b03741
键长:通常指 X 与 Y 之间的距离(而非 H 与 Y),经典氢键的 X…Y 距离一般为 2.5-3.2 Å(1Å=10⁻¹⁰m),且小于 X 和 Y 的范德华半径之和;
键角:指 X-H…Y 的夹角,理想情况下接近 180°,此时 Y 的孤电子对与 X-H 的反键轨道重叠最佳,键角越小,氢键强度越弱;
键能:破坏 1mol 氢键所需的能量,经典氢键的键能为 1-40 kcal/mol(1 kcal/mol≈4.18 kJ/mol),其中:
弱氢键:1-5 kcal/mol(如 C-H…O);中强氢键:5-20 kcal/mol(如 O-H…O);强氢键:20-40 kcal/mol(如 [F-H-F]⁻,键能约 40 kcal/mol,接近弱共价键)。
图4. DFT计算键长。10.1002/chem.202200755
氢键无法通过光学显微镜直接观察,需借助专门的实验技术和计算方法,从结构、光谱、能量三个维度综合分析。
XRD是最常用的方法之一。当 X 射线穿过晶体时,会被分子散射,通过分析散射图案可重构分子的三维结构,直接观察到 X-H…Y 的空间位置(键长、键角)。例如 DNA 双螺旋的结构,就是通过 XRD 首次确定了碱基对间的氢键;
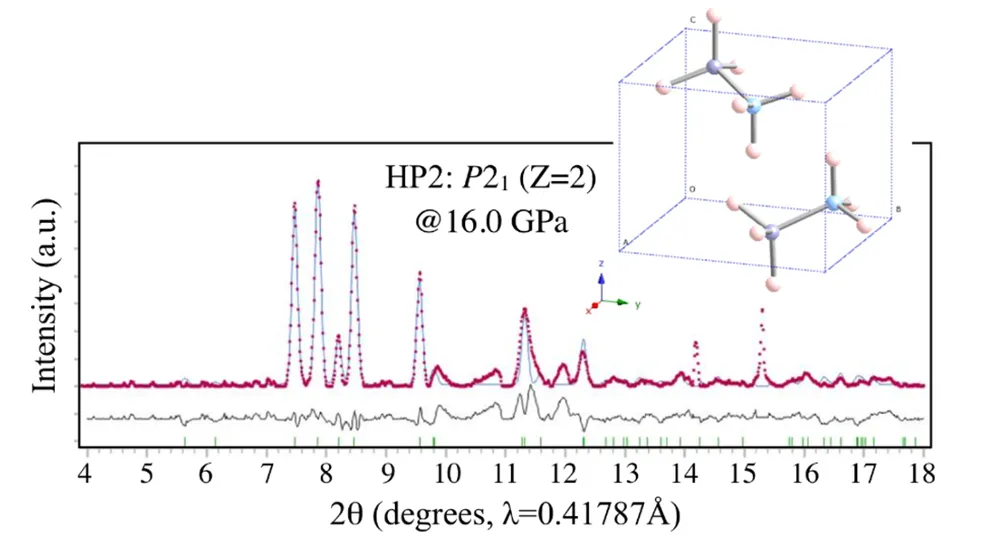
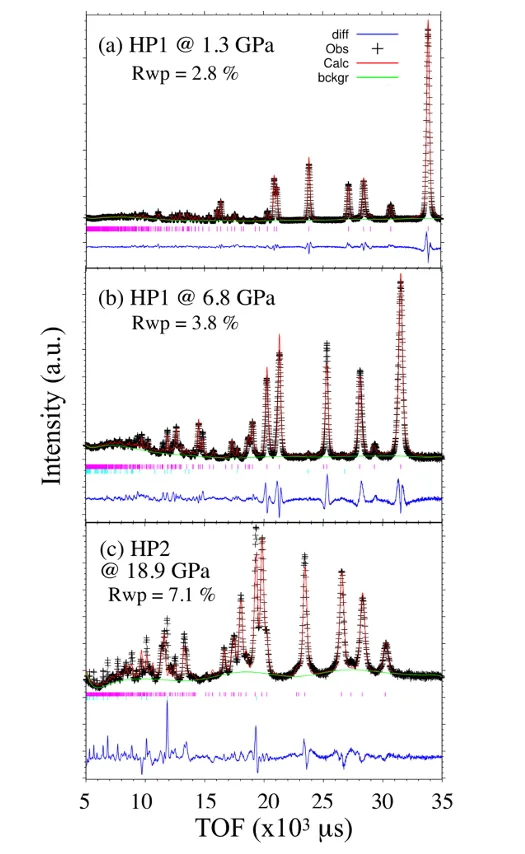
图5. HP2在16GPa下XRD图样的Rietveld精修。DOI:10.1021/acs.inorgchem.0c03345
比 XRD 更适合定位 H 原子。X 射线对轻原子(如 H)的散射能力弱,而中子对 H 的散射清晰,可精准测量 H 与 Y 的距离,常用于分析强氢键(如 [O-H-O]⁻)的质子位置;
图6. ND3BD3 的中子衍射图的Rietveld精修。DOI:10.1021/acs.inorgchem.0c03345
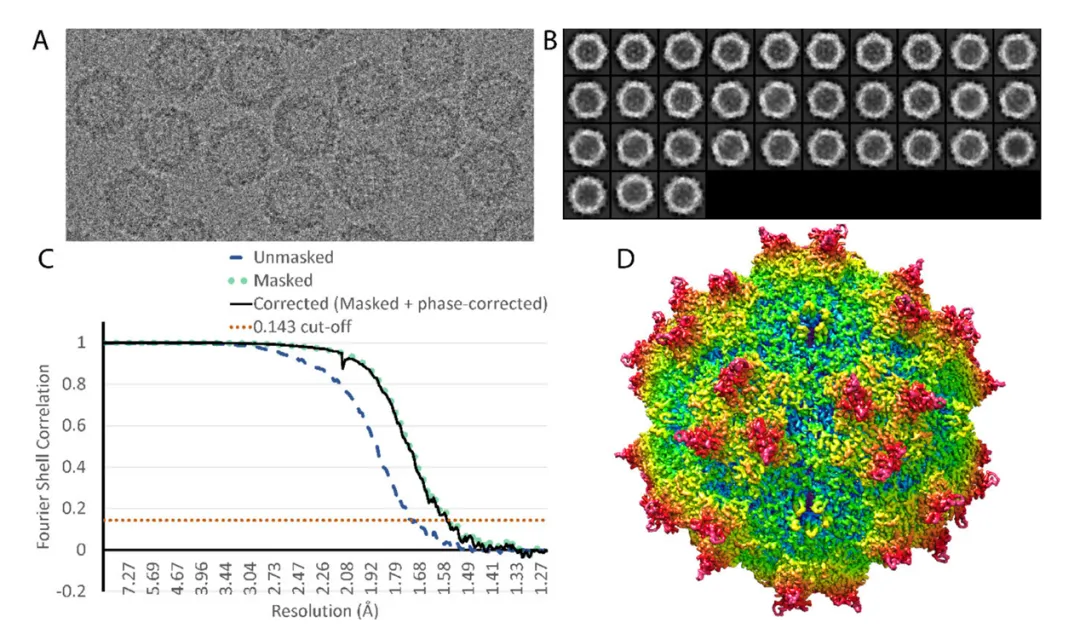
适用于生物大分子(如蛋白质、RNA)。通过快速冷冻样品,保持分子的天然构象,再通过电镜成像重构结构,可观察到蛋白质活性位点与底物间的氢键。
图7. 腺相关病毒 (AAV-DJ) 冷冻电镜重构结构。DOI:10.3390/v12101194
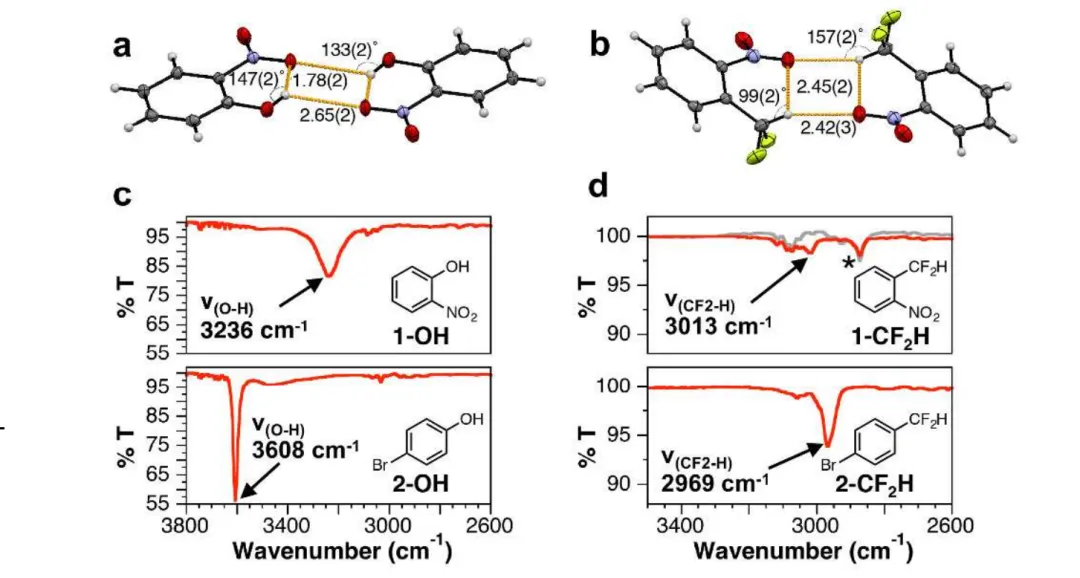
氢键会显著改变 X-H 的伸缩振动频率。例如游离的 O-H 伸缩振动峰在 3600 cm⁻¹ 左右,形成氢键后,O-H 键被 “拉弱”,振动频率降低(红移),峰形变宽 —— 氢键越强,红移越明显(如强氢键的 O-H 峰可红移至 3000 cm⁻¹ 以下);
图8. 100 mM浓度的1-OH、对溴苯酚(2-OH)、1-CF2H和对溴-α,α-二氟甲苯(2-CF2H)在CCl4中的红外光谱。10.1021/jacs.7b04457
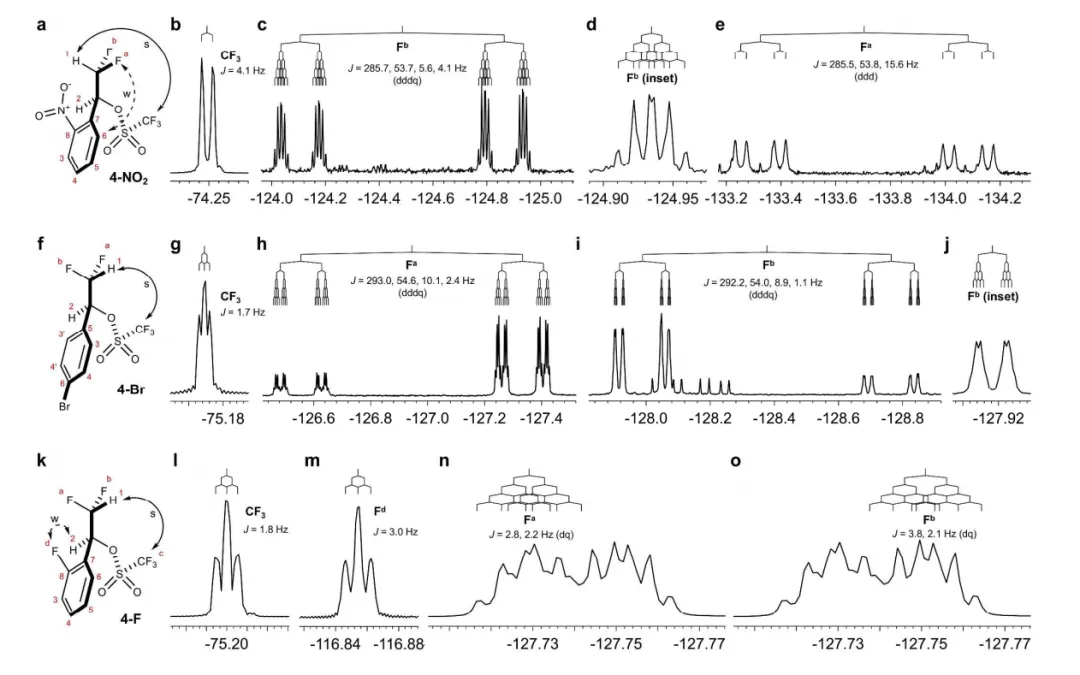
通过氢核(¹H)的化学位移判断氢键。游离的 H 化学位移较小(如甲醇的 O-H 在 δ=4.0 左右),形成氢键后,δ+H 的电子云密度降低,化学位移向低场移动(δ 值增大,如与 O 结合的 O-H 可移至 δ=10-15);此外,NMR 还可通过 “核 Overhauser 效应(NOE)” 测量 H 与 Y 的距离,验证氢键存在;
图9. 4-NO2(a)、4-Br(f)和4-F(k)核磁共振构象分析。10.1021/jacs.7b04457
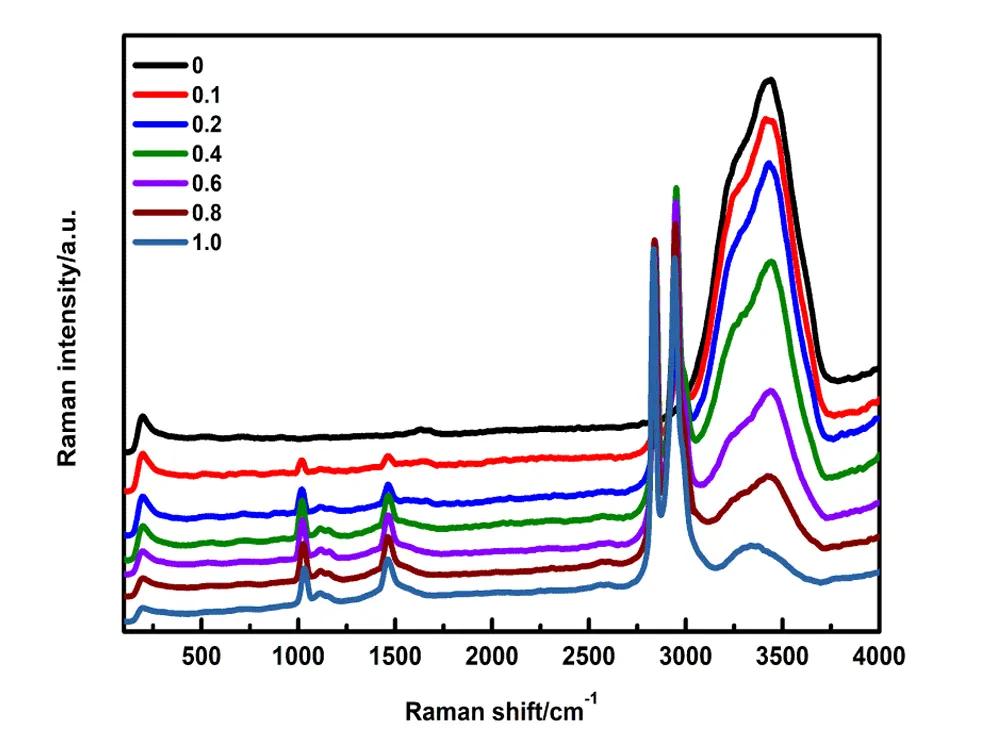
与 IR 互补,适用于水溶液等 IR 难以分析的体系。氢键会改变分子的极化率,导致拉曼峰的位移或强度变化,可用于研究水的氢键网络动态。
图10. 甲醇-水二元溶液随着甲醇体积分数变化的拉曼光谱。DOI:10.1016/j.saa.2019.117488
当实验无法直接测量时,如酶活性位点的瞬时氢键,计算模拟可作为重要补充:
基于量子力学原理,计算分子的电子结构。常用方法包括 “密度泛函理论(DFT)” 和 “从头算(ab initio)”,可计算氢键的键能、电荷分布、轨道相互作用,甚至预测氢键的红外光谱;
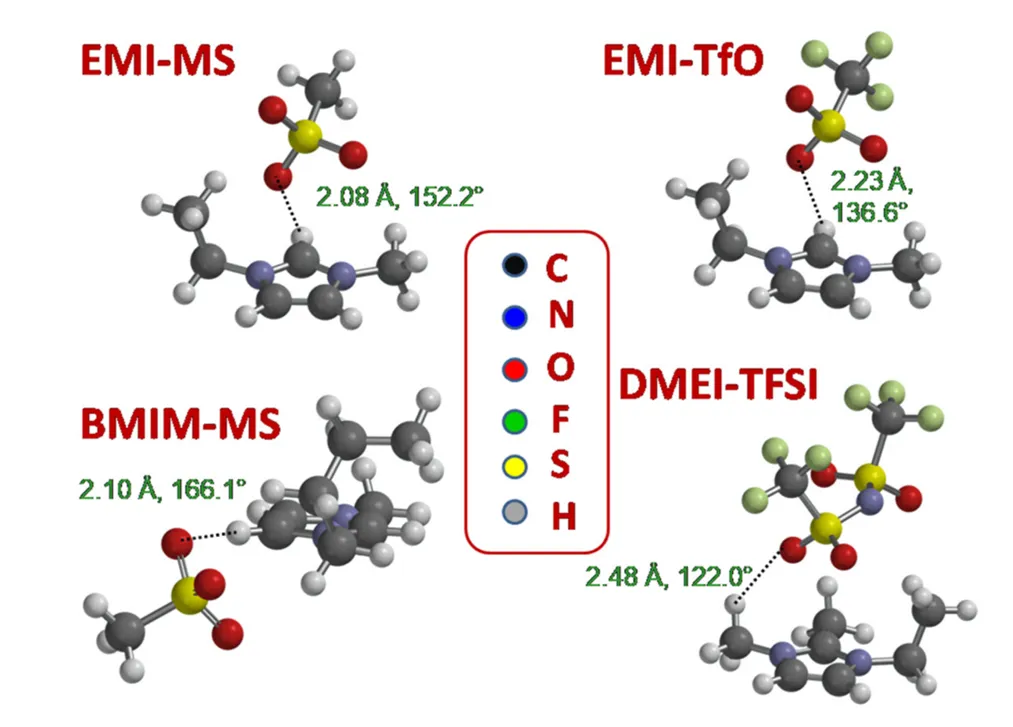
图11. 四种离子对的 DFT 计算。DOI: 10.3390/ijms22116155
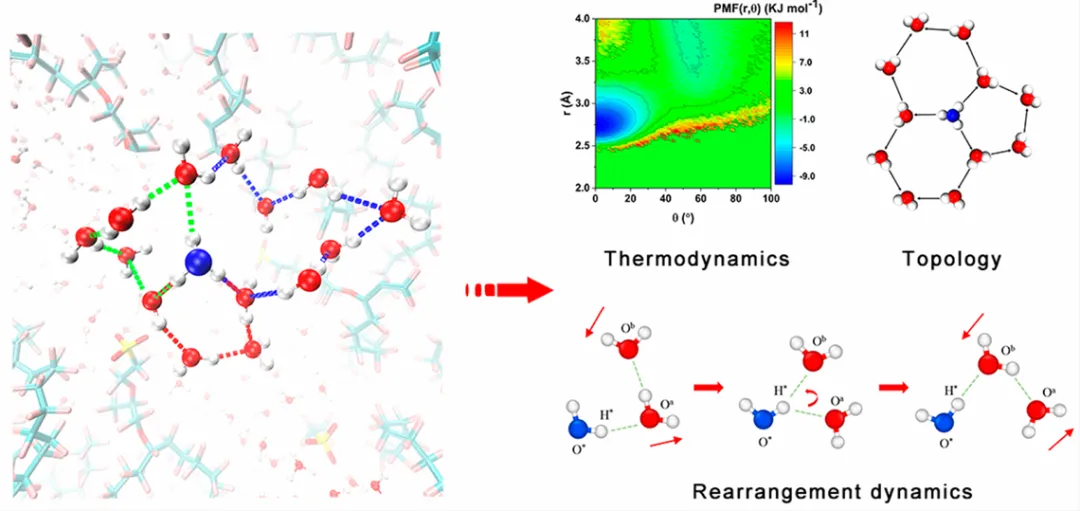
基于经典力学,模拟大量分子的动态行为。例如通过 MD 模拟可观察水溶液中氢键的 “断裂 – 重组” 过程(时间尺度可达纳秒至微秒),分析氢键的寿命和网络稳定性;
图12. 通过分子动力学模拟 Nafion 中氢键网络的演变。DOI: 10.1021/acs.macromol.2c02106
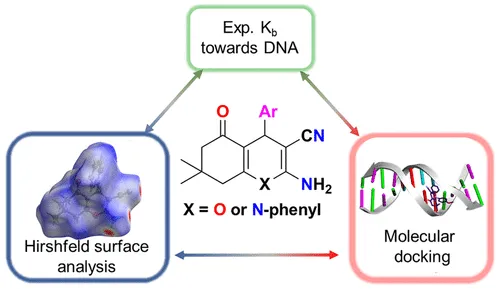
在药物研发中,通过对接计算预测药物分子与靶点蛋白的结合模式,分析氢键的数量和强度,筛选高亲和力的药物候选物。
图13. 杂环 rac-2-Amino-3-carbonitrile 衍生物的经典分子间氢键基序:连接 Hirshfeld 表面分析、CT-DNA 结合亲和力和分子对接。DOI: 10.1021/acs.cgd.1c01514
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!