

说明:本文华算科技从理论计算的角度,系统介绍反应位点(Reaction Sites)的基本概念、核心原理及其在化学催化中的研究进展。
内容涵盖反应位点的定义、热力学特性、计算方法(如密度泛函理论、从头算方法和其他高级工具)以及在酶工程、异相催化和药物设计中的重要性。
读者可通过本文了解反应位点的独特机制、模拟技术的关键作用,以及其在先进催化系统设计中的潜力,为计算化学、材料科学和生物工程的创新研究提供理论支持和实践指导。


什么是反应位点?

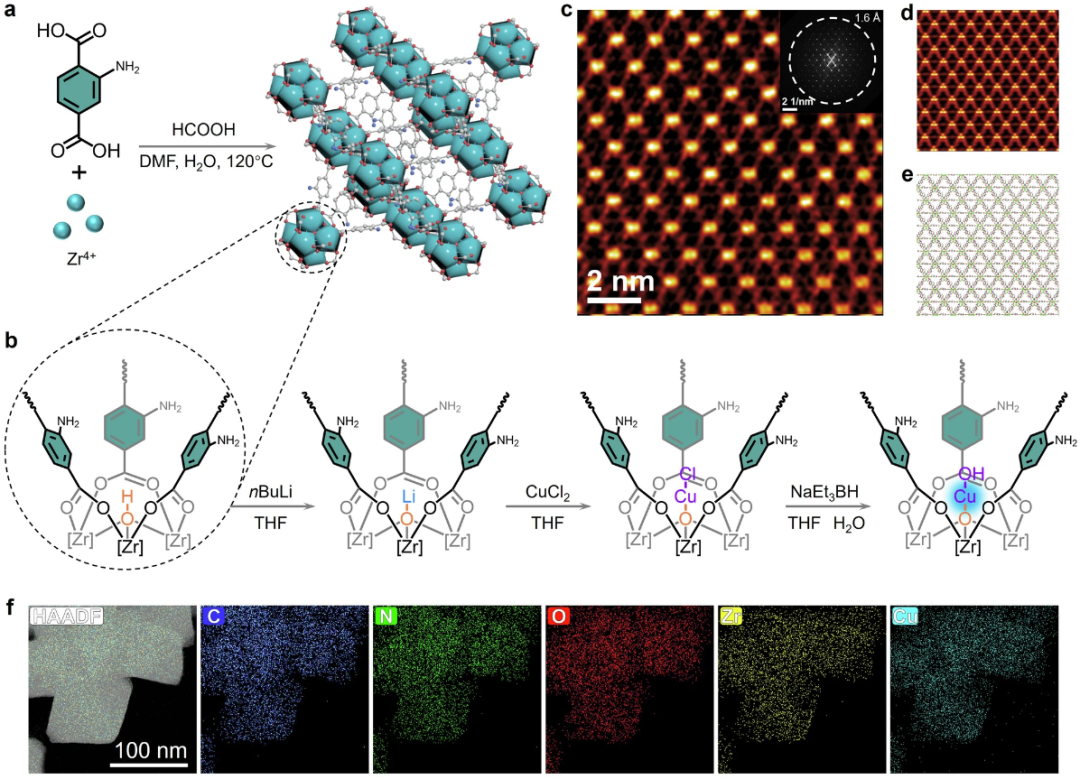
DOI: 10.1038/s41467-024-53483-z
反应位点是指催化剂或酶分子中直接参与化学反应、降低反应活化能的特定区域或原子簇。这些位点通常由氨基酸残基、金属离子或表面原子组成,通过与底物形成过渡态,实现高效的催化过程。
在酶催化中,反应位点往往位于蛋白质的腔体中,提供精确的立体环境和化学功能;在异相催化中,反应位点可能为纳米颗粒的边缘、台阶或缺陷位。
反应位点的核心原理源于过渡态理论,其中位点稳定中间体,降低反应势垒,从而加速反应速率。
传统实验方法如X射线晶体学可表征位点结构,但理论计算方法在揭示动态行为和电子转移机制方面具有独特优势。这些计算工具不仅能预测位点的几何构型,还能评估其对反应选择性和效率的影响,推动从分子水平到宏观应用的催化设计创新。
反应位点的理论计算方法
理论计算在反应位点研究中扮演关键角色,用于预测位点结构、反应机制和性能优化。以下介绍主要计算方法及其在反应位点中的应用。
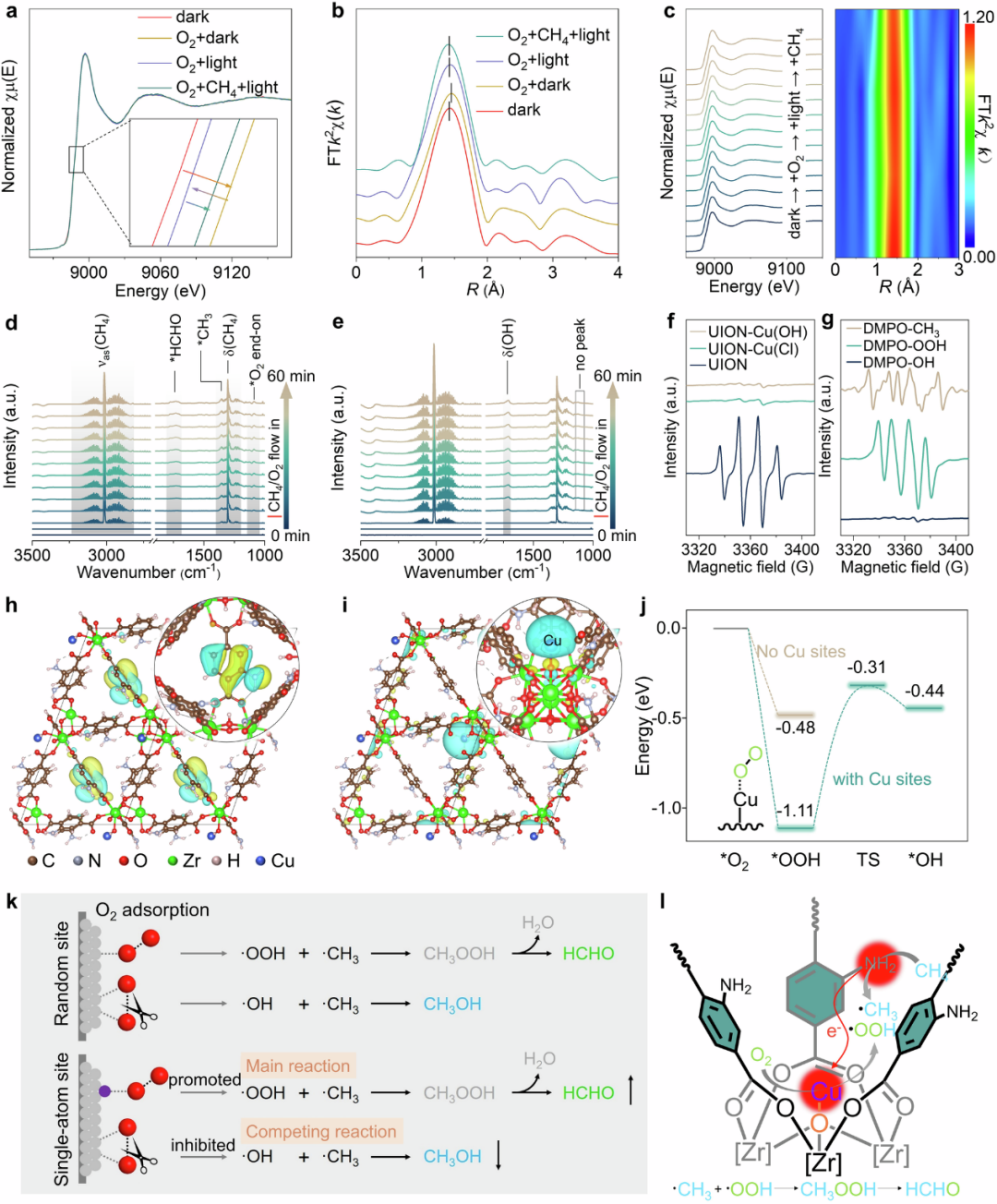
DOI: 10.1038/s41467-024-53483-z
密度泛函理论基于量子力学,计算反应位点的电子结构、结合能和反应势垒。其核心优势是无需经验参数,直接从电子层面预测原子间相互作用和催化活性。
例如,DFT用于模拟催化表面上的反应机制,揭示反应位点如何通过表面原子排列稳定过渡态,形成低能量路径,从而实现选择性催化反应。
该方法特别适用于异相催化体系,如金属纳米颗粒的边缘位点,在评估吸附能和活化能时表现出色。
应用:DFT在反应位点工程中应用广泛,如预测金属表面缺陷位对活性的影响和异相催化中的位点识别。挑战在于计算复杂体系的精度,需通过高级泛函和嵌入方法提升准确性,结合实验数据校正。
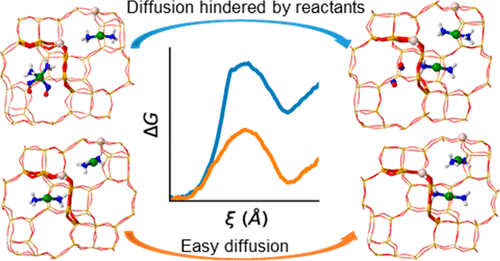
DOI: 10.1021/jacsau.1c00337
从头算方法如Hartree-Fock(HF)和耦合簇(CCSD)理论,从基本物理原理出发计算波函数,提供高精度电子相关描述,适用于小分子反应位点的精确模拟。其优势在于无经验参数的纯理论基础,能准确捕捉多参考态和强相关效应。
例如,[Cu(NH3)2]+复合物在菱沸石结构的两个相邻空腔之间的迁移,包括其他反应物分子(NO、O2、H2O 和NH3),在初始和最终空腔中的迁移为使用ab initio 分子动力学 (AIMD) 模拟结合增强的采样技术来描述从一个笼子到另一个笼子的跳跃事件。
我们发现这种扩散仅受到过量 NH3的存在显着阻碍或初始空腔中的NO,因为两种反应物都与[Cu(NH3)2]+稳定的中间体形成,这些中间体体积太大而无法穿过连接空腔的8环窗口。O2的存在极大地改变了NO与Cu+的相互作用。
其他不常见的方法包括量子力学/分子力学混合(QM/MM)和自动化反应路径搜索工具。这些方法扩展了传统计算的适用范围,提供更全面的位点识别和机制探索。
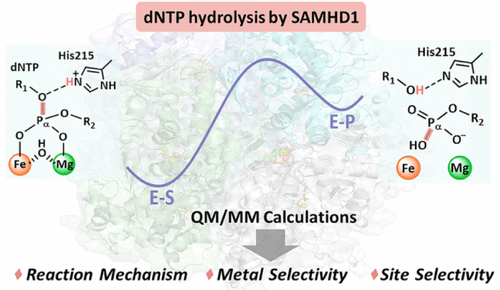
DOI: 10.1021/acscatal.5c01682
QM/MM方法:将量子力学处理反应位点核心区,分子力学模拟外围环境,适用于生物催化体系。例如,采用分子动力学(MD)模拟和量子力学/分子力学(QM/MM)计算来研究由两个金属离子介导的dATP水解的机理细节。
自动化机制搜索:如基于图论的算法,从蛋白质数据库(如PDB)搜索潜在位点,或使用过渡态搜索工具自动生成反应网络。这些工具加速了高通量筛选,如在异相催化中识别新型缺陷位点,但需高性能计算支持。
结论
反应位点作为催化反应的核心,通过精确的结构和动态机制实现高效转化,成为化学和生物工程的焦点。理论计算方法——密度泛函理论、从头算方法及其他高级工具——为反应位点的机制解析和优化提供了强大工具。
这些方法通过电子结构建模、波函数计算和混合模拟策略,显著推进了酶工程、异相催化和能源转换中的应用。随着计算技术和算法的进步,如自动化工具的集成,反应位点的设计将进一步加速,为绿色化学和可持续技术提供新机遇。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???