

摘要
X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)是一种使用电子谱仪测量X射线光子辐照时样品表面所发射出的光电子的方法,在化学、材料学、化学工程等领域都有着广泛的应用和重要意义,对于深入理解材料表面性质以及开发新材料具有不可替代的作用。本文介绍了XPS技术的功能、特点和应用领域,简要分析了XPS样品和谱图的处理方法,并对XPS在能源材料领域的前沿应用进行了举例说明。
目录
1. XPS基本知识
1.1 XPS概述
1.2 XPS技术的起源
1.3 XPS的原理
1.4 X射线光电子能谱仪的结构
2. XPS技术简介
2.1 XPS技术的功能
2.2 XPS技术的特点
2.3 XPS技术的应用
3. XPS测试
3.1 XPS样品处理
3.2 XPS谱图
4. XPS数据处理
4.1 XPS分析软件及数据库
4.2 Avantage软件处理数据
4.3 非典型图谱处理
5. XPS在能源材料领域的前沿应用
5.1 原位电化学XPS
5.2 XPS深度分析
6. XPS的局限性
结语
参考书籍
参考文献
1. XPS基本知识
1.1 XPS概述
XPS是一种常用的显微分析技术,常常和俄歇电子能谱技术(AES)配合使用,其发展历程可以追溯到20世纪初光电效应的发现。XPS可以比AES更准确地测量原子的内层电子束缚能及其化学位移。XPS可以提供分子结构和原子价态方面的信息,揭示各种化合物的元素组成和含量、化学状态、分子结构、化学键等信息。因为入射到样品表面的X射线束是一种光子束,所以XPS测试对样品的破坏性非常小,是一种无损检测手段。
处于原子内壳层的电子结合能较高,要把它激发出来需要能量较高的光子,以Mg或Al作为阳极材料的X射线源得到的光子能量分别为1253.6 eV和1486.6 eV,这个范围内的光子能量足以把大部分轻原子的核外电子激发出来。由于这些电子的结合能值各不相同,而且各元素之间相差很大,容易识别。因此,通过分析激发电子的结合能可以鉴定样品中的化学元素。除了不同元素的同一内壳层电子的结合能各有不同外,原子的内壳层电子的结合能还与该原子的化学结合状态及其化学环境有关。随该原子所在分子和键合状态不同,该内壳层电子的光电子峰会有位移,称为化学位移。这是由于内壳层电子的结合能除了受原子核电荷的影响外,还受周围价电子的影响。通过对化学位移的考察,XPS成为研究电子结构、高分子结构和链结构的有力工具。
1.2 XPS技术的起源
1887年,Heinrich Rudolf Hertz发现了光电效应,1905年,Albert Einstein对光电效应进行了理论解释,并为此获得了1921年的诺贝尔物理学奖。1907年,P.D. Innes用伦琴管、亥姆霍兹线圈、磁场半球(电子能量分析仪)和照像平版做实验来记录宽带发射电子和速度的函数关系,他的实验事实上记录了人类第一条X射线光电子能谱。
XPS的研究由于战争而中止,直到战后,Kai Siegbahn和他的团队在瑞典乌普萨拉大学开发出了现代XPS技术,并于1954年获得了氯化钠的首条高分辨X射线光电子能谱,显示了XPS技术的强大潜力。1967年之后的几年间,Siegbahn就XPS技术发表了一系列学术成果。在与Siegbahn的合作下,美国惠普公司于1969年制造了世界上首台商业单色X射线光电子能谱仪。1981年,Siegbahn研制出第一台高分辨率电子能谱仪,并在当年获得了诺贝尔物理学奖。
1.3 XPS的原理
当一束光子辐照到样品表面时,光子可以被样品中某一元素的原子轨道上的电子所吸收,使得该电子脱离原子核的束缚,以一定的动能从原子内部发射出来,变成自由的光电子,而原子本身则变成一个激发态的离子。
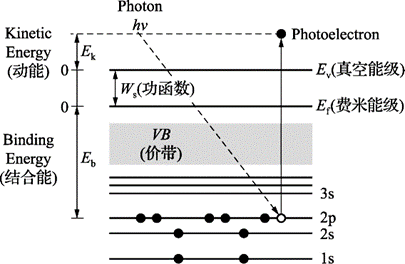

图1.1 样品受激发后光电子的发射过程
根据爱因斯坦光电发射定律有:
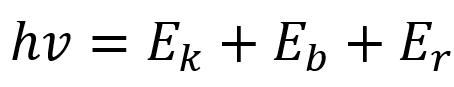
式中,hv为X射线源光子的能量;Ek为出射的光电子动能;Eb为特定原子轨道上的结合能;Er是原子的反冲能量,很小,可以忽略。
因此,可得到下述关系:
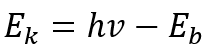
当固定激发源能量hv时,其光电子的能量Ek仅与元素的种类和所电离激发的原子轨道有关。因此,我们可以根据光电子的结合能定性分析物质的元素种类。
通过测量样品中各个元素光电子结合能的大小来鉴别样品表面元素的化学组成、状态及含量,从而进行定性、定量分析。
1.4 X射线光电子能谱仪的结构
X射线光电子能谱仪由X射线激发源、真空系统、能量分析器、电子倍增器和控制系统等构成,各部分的作用如下:
a,X射线激发源。常用的激发源有Mg Kα(光子能量为1253.6 eV)和Al Kα (光子能量为1486.6 eV)。原始的X射线线宽约0.8 eV,经单色化处理后,线宽可降低至0.2 eV,并可以消除X射线中的杂线和韧致辐射。但处理后的X射线的强度会大幅度下降。
b,真空系统。XPS必须在高真空环境下测试,主要有两方面原因。首先,XPS是一种表面分析技术,分析深度为几个纳米,如果分析室的真空度很差,在很短的时间内试样表面就可能会被真空中的残余气体分子所覆盖,从而难以获取样品表面的真正信息。其次,由于光电子的信号和能量都非常弱,如果真空度较差,光电子很容易与真空中的残余气体分子发生碰撞作用而损失能量,甚至难以检出。通常超高真空系统真空度优于10-9 Torr。
c,能量分析器。X射线光电子的能量分析器有两种,半球形能量分析器和筒镜形能量分析器,用于精确测定出射的光电子动能。
d,电子倍增器。光电子能谱仪中被检测的电子流非常弱,所以现在多采用电子倍增器加计数技术来放大电子流。
e,控制系统:能量分析器和电子倍增器系统是由计算机控制的,经过数据采集和处理系统后显示出结果。
2. XPS技术简介
2.1 XPS技术的功能
a,定性分析。根据所测得谱的位置和形状来得到有关样品的组分、化学态、表面吸附、表面态、表面价电子结构、原子和分子的化学结构、化学键合情况等信息。元素定性的主要依据是组成元素的光电子线的特征能量值。XPS能够分析除H,He以外的所有元素,灵敏度约0.1 At%。此外,能够通过观测化学位移来判断原子氧化态、原子电荷和官能团等信息。化学位移信息是利用XPS进行原子结构分析和化学键研究的基础。
b,定量分析。以能谱中各峰强度的比率为基础,把所观测到的信号强度转变成元素的含量,即将谱峰面积转变成相应元素的含量,多采用元素灵敏度因子法,该方法利用特定元素谱线强度作参考标准,测得其它元素相对谱线强度,求得各元素的相对含量。XPS定量分析除了可以计算不同元素的相对原子浓度,对同一种元素在不同化学态下的原子相对浓度也可进行分析。但是同一元素不同化学态下原子的结合能峰位很接近,会叠加在一起形成宽峰。这时要想通过解析这些原子的峰强度比来获得它们的相对含量,就要将宽峰分解成组成它的各个单峰,即去卷积。
c,深度剖析。为了获得深度大于10 nm的元素化学信息,可以在XPS设备的分析室用惰性气体离子轰击,对样品表面进行刻蚀。使用Ar离子枪对样品表面进行溅射剥离,通过控制合适的溅射强度及溅射时间,将样品表面刻蚀到一定深度,然后进行取谱分析。为了获得准确的溅射深度,一般采用与被测样品相近或相同的标准物质校准溅射速率,从而根据溅射时间计算得到校准后对应元素分布的溅射深度。
d,角分辨电子能谱分析。改变样品表面与入射光束间的角度,即可改变入射光的检测深度,使得检测深度变浅,这样来自最表层的光电子信号相对较深层的会大大增强。利用这一特性,可以对超薄样品膜表面的化学信息进行有效地检测,从而研究超薄样品化学成分的纵向分布。但需要注意的是,这种方法仅适用于衬底上覆盖层均匀且厚度小于10 nm的薄膜。
2.2 XPS技术的特点
a,高灵敏度。可以检测到非常低浓度的元素,可以达到0.1 At%级别。
b,高分辨率。提供非常精确的能量分辨率,可以区分非常接近的能级。
c,高测试范围。除了H和He之外,XPS可以分析元素周期表中的所有元素。
d,检测化学位移。化学位移可以用于材料中结构分析和化学键研究。
e,无损检测。XPS是一种非破坏性的表面分析技术,可以在不破坏样品的情况下获取材料表面的化学信息。
f,深度剖析。通过Ar离子溅射剥离样品表面的方法,可以探测样品深度方向上的化学状态变化。
g,小于10 nm薄膜分析(角分辨)。XPS可以对非常薄的薄膜进行分析,甚至可以达到纳米级别的分辨率。
h,元素化学态分布(XPS成像技术)。XPS可以通过成像技术来观察样品中元素的分布情况,从而了解元素的化学态分布。
2.3 XPS技术的应用
a,催化和表面科学:用于研究材料的表面化学成分、吸附物、价电子结构等。可以帮助了解材料的表面性质,优化材料的性能,并研究材料的表面反应和催化机制。
b,界面和薄膜:有助于分析界面和薄膜的形成机制和性能,并研究界面和薄膜之间的相互作用。
c,表面工程:在材料表面改性、涂层制备、表面摩擦磨损性能评估等方面的应用,光电子能谱分析可以用于评估材料在腐蚀与防护过程中的性能,包括耐蚀性、耐磨性、耐热性等。
d,生物医学:XPS可以用于研究生物材料的表面化学成分和表面性质。它可以对生物材料的相容性、附着性和生物活性进行研究。
e,环境和能源:研究环境污染物的表面化学成分和表面性质,了解污染物的来源、迁移和转化,并研究环境污染和能源转化的机制。
f,电池及储能技术:分析正负极材料的表面化学状态,评估电池的性能和稳定性;研究电池材料的表面反应和电化学界面特性。
3. XPS测试
3.1 XPS样品处理
XPS作为一种高灵敏度的表面分析方法,测试结果难免会受到各种因素的影响,因此对样品的要求较高。XPS测试方法一般为送样测试,送样前需对样品进行前处理以满足以下要求:
a,尺寸不大于5×5 mm2。XPS为微区测试,样品尺寸不需要很大。
b,干燥,不能释放气体。由于XPS样品室真空度极高,未完全干燥或会释放气体的样品将导致真空度不达标而无法进行测试。通常送样前需对样品进行高温真空干燥(> 60℃)或冷冻干燥24小时上。
c,样品表面导电。不导电的样品会积累电荷产生荷电,从而影响光电子的发射。
d,表面平整,清洁。待测样品表面应做脱脂处理,不能用手或手套接触,避免在空气中长时间存放。
3.2 XPS谱图
XPS谱分为全谱和精细谱(高分辨谱)。全谱分析一般用来确认样品中的元素种类和含量(H和He除外)。但是全谱分析所得到的信号较为粗糙,只是对元素进行粗略的扫描,确定元素有无以及大致含量。对于含量较低的元素而言,信噪比很差。通常,全谱分析只能得到表面组成信息,得不到准确的元素化学态和分子结构信息等。下图为通过全谱分析不同碳化温度制备的氮掺杂碳纳米纤维中氮的含量,三种样品的氮含量分别为13.8 At%、8.1 At%和4.9 At%。
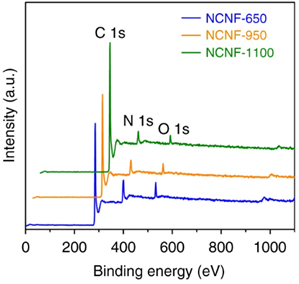
图3.1 全谱用于分析元素含量
除了全谱外,XPS测试过程中常常对某些元素进行精细分析,精细分析获得的谱图称为精细谱。通过分析精细谱的峰位可以判断元素的化学状态。此外,可以通过对比处理前后样品表面元素的化学位移变化,来说明样品的表面化学状态或者是样品表面元素之间的电子相互作用。一般某种元素失去电子,其结合能会向高场方向偏移,得到电子反之(例外元素:如钴等)。下图所示是Pt和Pd形成合金后其表面电子结构的变化,从图中可以看出,形成Pt1Pd3之后,Pd 3d向低结合能偏移,Pt 4f向高结合能偏移,说明Pd得到电子,Pt失去电子,也就是说形成合金后,Pt上的电子部分转移给Pd。
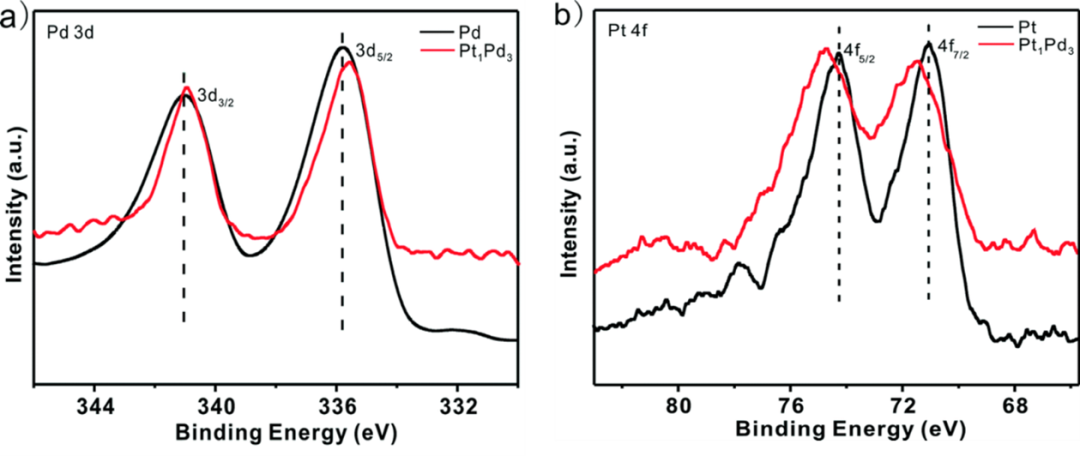
图3.2 Pd和Pt精细谱用于分析合金中电子状态
4. XPS数据处理
4.1 XPS分析软件及数据库
XPS常用的数据分析软件包括Thermo Avantage(下文简称为Avantage)、Casa XPS和XPS Peak等,其中Avantage软件自带一系列数据处理工具,包括峰值拟合、基线校正、光谱平滑等,而且包含了广泛的元素和化合物的数据库,是分析XPS数据的利器。下文将介绍Avantage软件的使用。
此外,XPS手册也常用来对照谱峰。网络数据库如http://www.lasurface.com/database/elementxps.php也可以用来进行分析。
4.2 Avantage软件处理数据
常规的XPS数据处理包括通过对全谱分析获得化合物中元素种类;对精细谱进行分析获得元素的化学态与结构。Avantage软件在全谱和精细谱分析中都提供了高度自动化的工具,可以轻松准确地对谱图进行分析。
4.2.1 软件界面
打开Avantage软件,界面如下。可以通过单击文件导入测试数据,也可以单击红色圆圈标记处打开自带数据库。
图4.1 Avantage软件界面
打开数据库,单击左上角按钮可以获得不同元素的精细谱数据库,内容包括结合能范围、标准图谱和测试注意事项等,因此建议在测试前先查阅数据库,获得合适的精细谱测试区间范围。
图4.2 Avantage数据库-以碳元素为例
4.2.2 文件导入
双击或者在Avantage软件导入测试文件,测试文件通常为.vms或.vgd格式。如下图所示,本次测试中全谱和精细谱都将一起导入。其中右下的图谱为全谱,其他三个图谱分别为Se 3d,C 1s和Ag 3d的精细谱。
图4.3 导入测试文件
4.2.3 更改X轴显示方式
首先左键点击红色圆圈标记处全选所有图谱,然后再右键菜单-显示选项中将横轴单位由动能改为结合能Binding Energy。
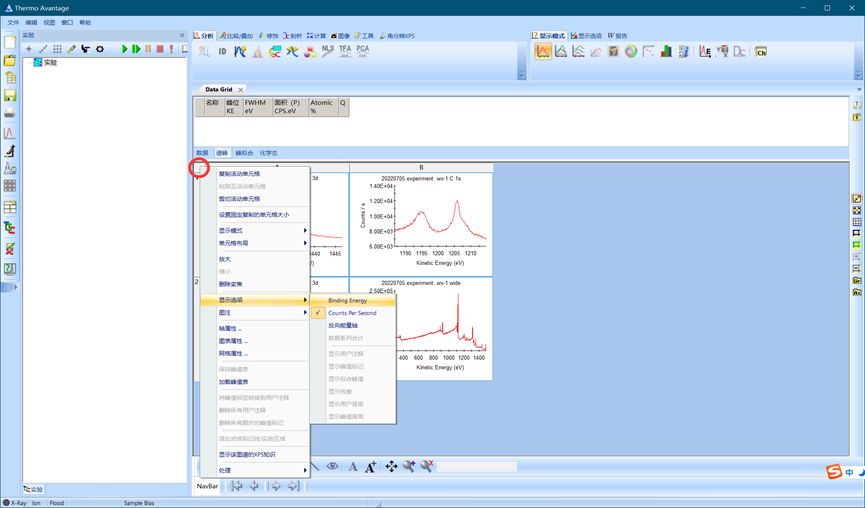
图4.4 更改横坐标为结合能
4.2.4 电荷移位
在XPS测试过程中,如果样品绝缘或导电性不好,经X射线辐照后,其表面产生的正电荷不能得到电子的补充而导致电荷积累,使测得的结合能比正常值要偏高。由于XPS分辨率高,微弱的荷电都会导致分析结果的偏差。因此需要通过碳峰对谱图进行校正。首先全选所有图谱,然后单击电荷移位按钮。
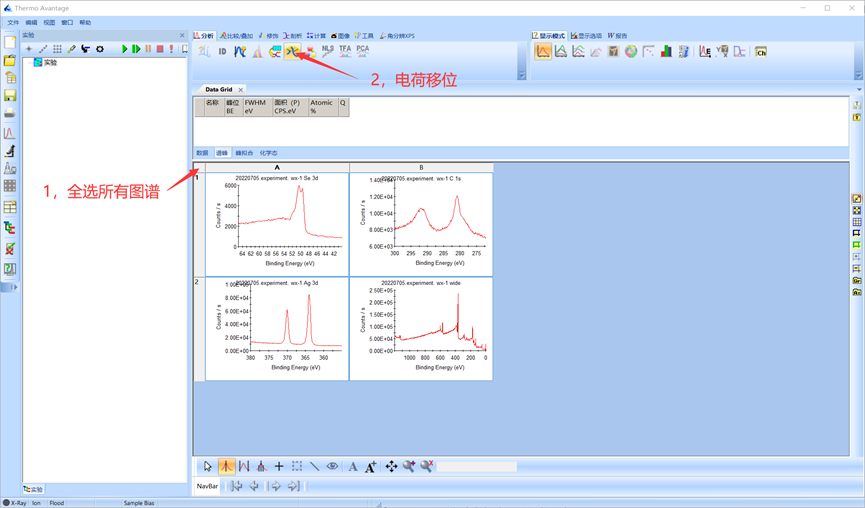
图4.5 单击电荷移位按钮
在电荷移位对话框中,首先勾选左下角“允许非整数通道宽度位移”然后输入位移量和位移方向,单击接受。通常以污染碳峰作为基准,不同仪器污染碳峰位置略有不同(284.8 eV 左右)。
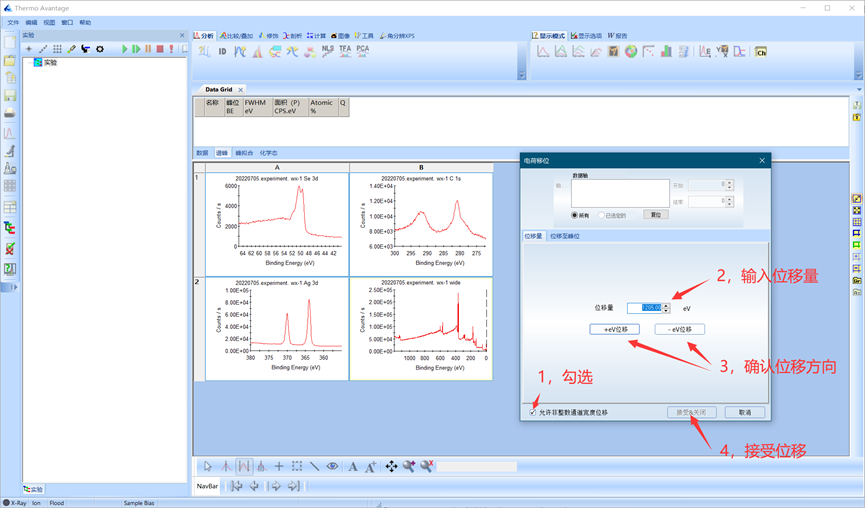
图4.6 手动电荷移位操作方式
此外,还有另外一种自动校正方式,在大多数情况下可以更快速、准确地进行校正。如下图所示,使用“位移至峰位”功能,依次点击C-1s,此时软件会自动计算位移值,确认位移值后单击位移并接受。
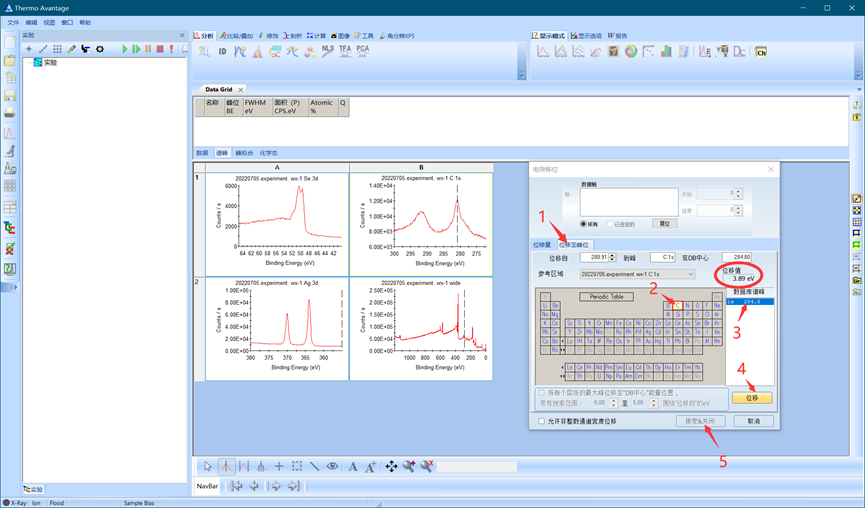
图4.7 第二种电荷移位方法
4.2.5 全谱分析
单击全谱,然后运行全谱自动识别。
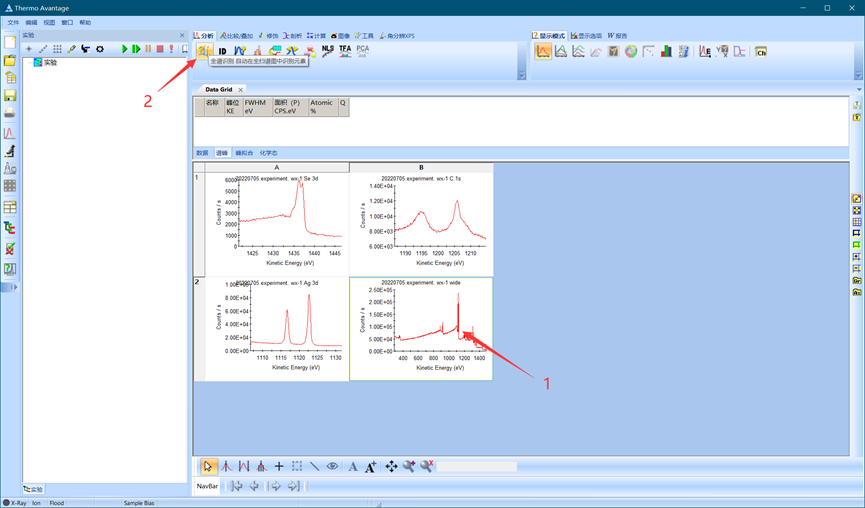
图4.8 运行全谱自动识别
运行后,软件自动生成元素比例。如有峰强度弱等现象,软件会在元素后标注???,此时可以放大图谱手动识别并删除误标的峰。
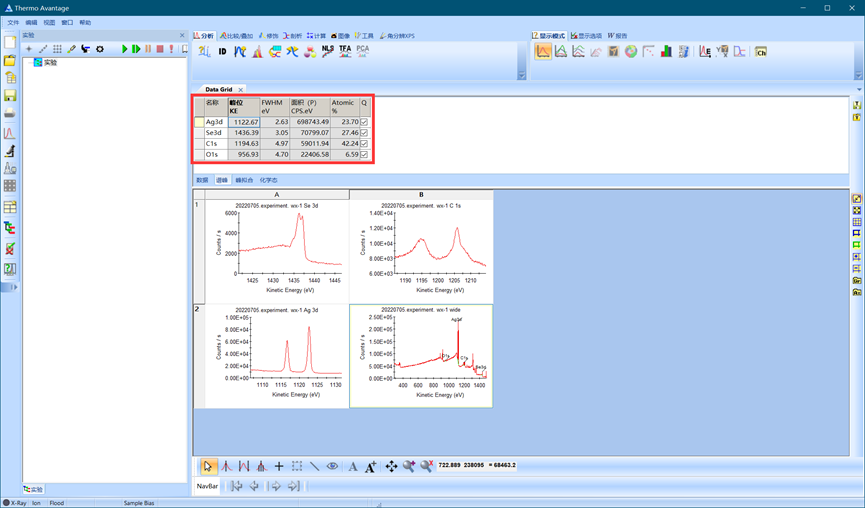
图4.9 全谱元素识别运行结果
如有峰未被识别到,可以放大图谱,使用手动峰位识别进行加峰。单击手动峰位识别后拖动识别范围,框选未标出峰,选择元素并添加。
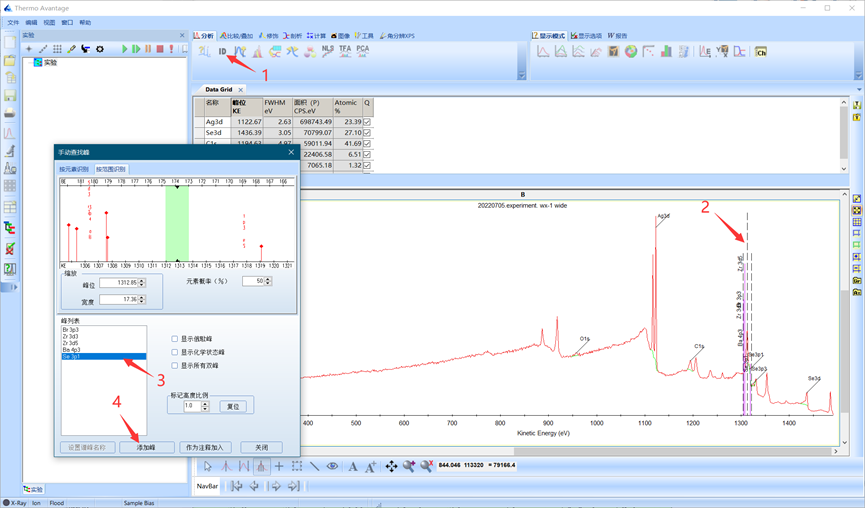
图4.10 全谱手动峰位识别与加峰
4.2.6 精细谱拟合-单峰拟合
元素的s轨道为单峰,可以自由添加单峰进行拟合。以C 1s为例。首先单击C 1s图谱,点击峰添加,框选合适的峰范围,添加背底,然后关闭对话框。
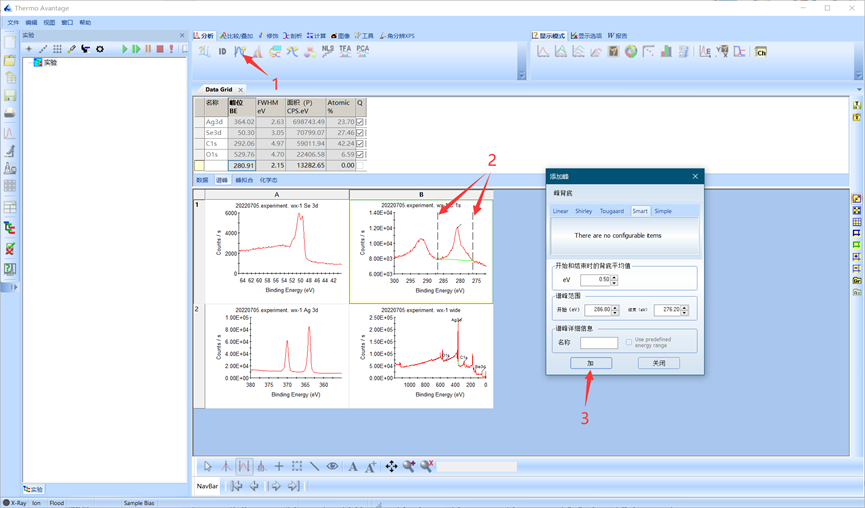
图4.11 添加背底
单击峰拟合按钮,此时会自动添加一个峰,切换到Add Fitted Peak菜单,移动游标,调节峰位和半峰宽至可能存在峰的位置,然后点击添加单个峰进行加峰。
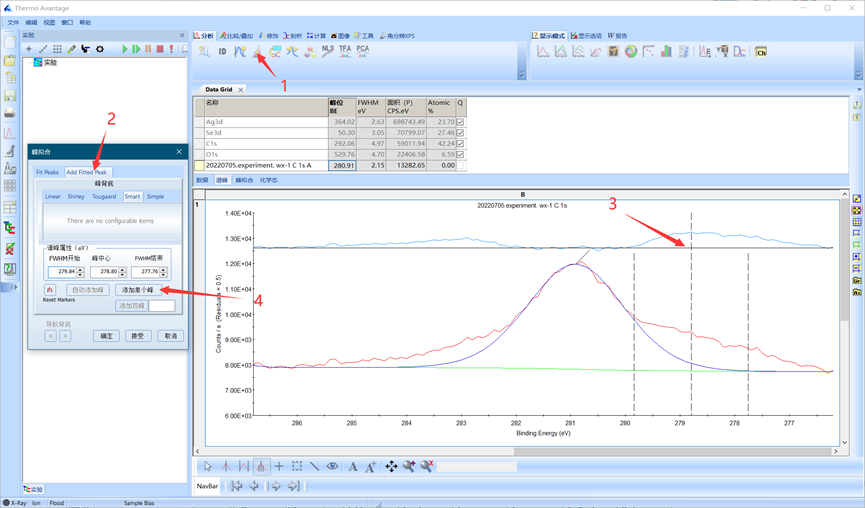
图4.12 添加单峰
本例所示C 1s可分为三个峰,添加三个峰后点击Fit Peaks按钮进行拟合。调节拟合参数后单击拟合这个层进行拟合,(拟合参数默认即可),多次拟合满足要求后点击接受并关闭对话框。
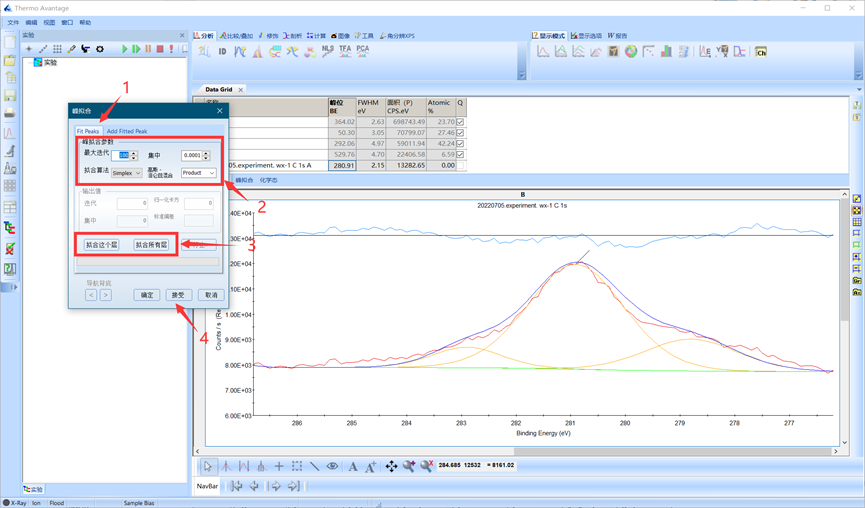
图4.13 运行单峰拟合
拟合后的图谱如下所示,本例所示C 1s分为的三个峰的峰位分别为284.77,282.57和286.72 eV,对照数据库判断其分别为C-C键、金属碳化物和C-O-C键。
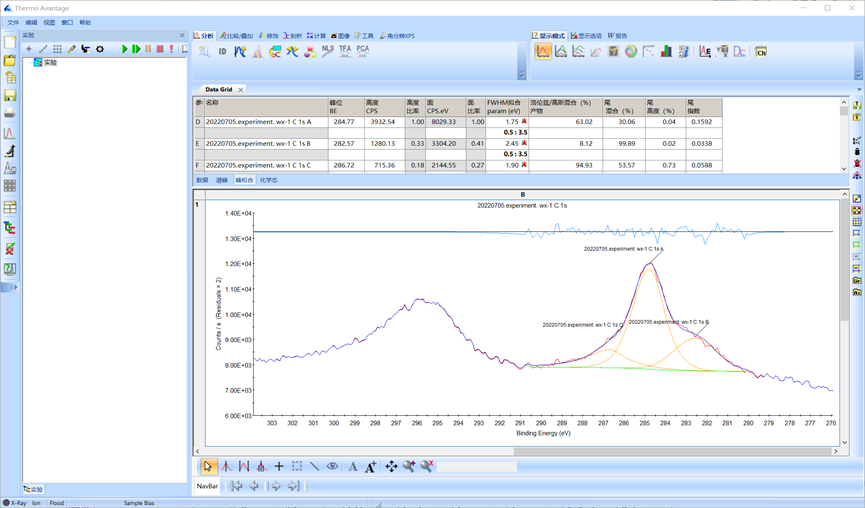
图4.14 单峰拟合输出结果
4.2.7 精细谱拟合-双峰拟合
元素的p,d,f轨道为分裂峰,显示在图谱上为双峰,双峰拟合结果比单峰更可靠。双峰的结合能差一般为定值,有时候可以反应元素价态;双峰的半峰宽相等,峰面积比为固定值,p,d,f轨道双峰的峰面积比分别为1:2,2:3和3:4。在双峰拟合时,Avantage软件会自动设定双峰的结合能差并限定半峰宽和峰面积比。
双峰拟合的步骤与单峰拟合相似,除了在手动加峰的过程中点击添加双峰。具体过程如下:加背底后单击峰拟合-Add Fitted Peak,此时会自动识别双峰,如果没有识别可以将三条虚线拖到峰强较高的峰上并手动输入(如下图步骤3,4)并点击添加双峰,后续拟合过程与单峰拟合一致。
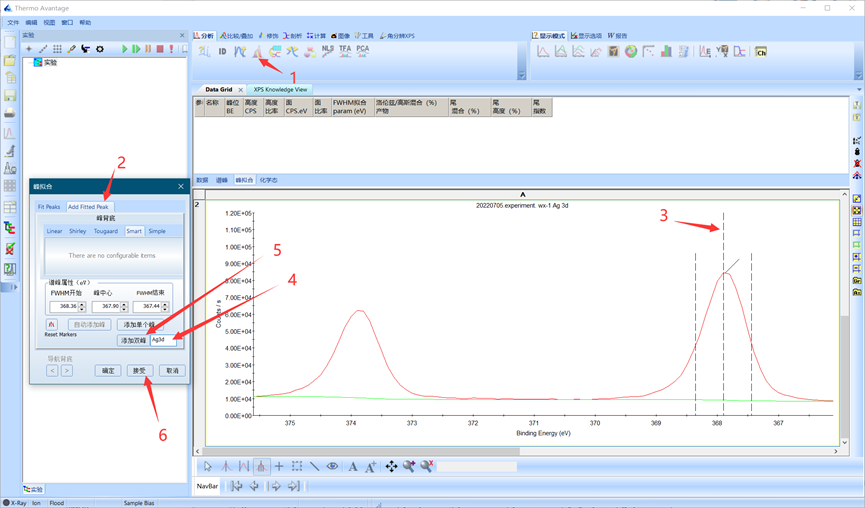
图4.15添加双峰
拟合完毕如下图所示,可以看到Avantage软件自动限制了双峰之间的结合能差,还限制了峰强、峰面积比例和半高宽,这最大限度地降低了我们分析XPS数据出错的可能性。
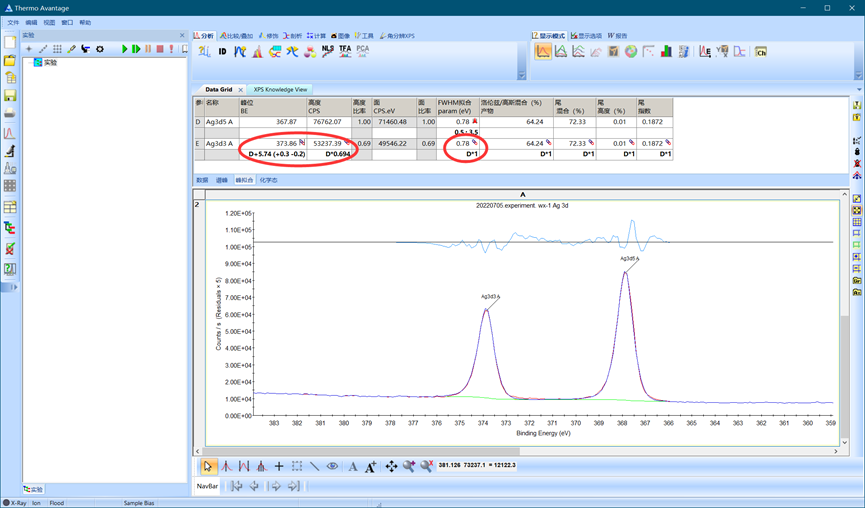
图4.16 双峰拟合结果
4.2.8 数据导出与绘图
在Avantage软件中选中图谱,右键-复制活动单元格即可将图谱中原始数据及峰拟合结果复制,粘贴到Origin等软件中即可进行绘图。绘图过程这里不再赘述。
图4.17 谱图数据导出
4.3 非典型图谱处理
在实际处理XPS图谱的过程中,情况往往会更复杂。这里举例几种常见元素XPS精细谱处理过程中可能会遇到的问题。其他元素请参考数据库和文献。
4.3.1 Fe 2p
由于常规X射线源(Al/Mg Kα1,2)并非是单色的,而是还存在一些能量略高的小伴线(Kα3,4,5和Kβ等),所以导致XPS中,除Kα1,2所激发的主谱外,还有一些小的伴峰(卫星峰)。
铁化合物分为高电子自旋和低电子自旋态,其中三价铁总是高电子自旋的,这会导致更多更复杂的多种卫星分裂峰。二价铁有的表现出高自旋,有的表现为低自旋,其中高自旋化合物(如FeO或FeCl2)的Fe 2p谱表现出复杂的多重态分裂,并具有卫星特征。低自旋化合物(如FeS2)的谱不显示多重态分裂或卫星特征。在分析过程中可以利用这个特性进行分析。
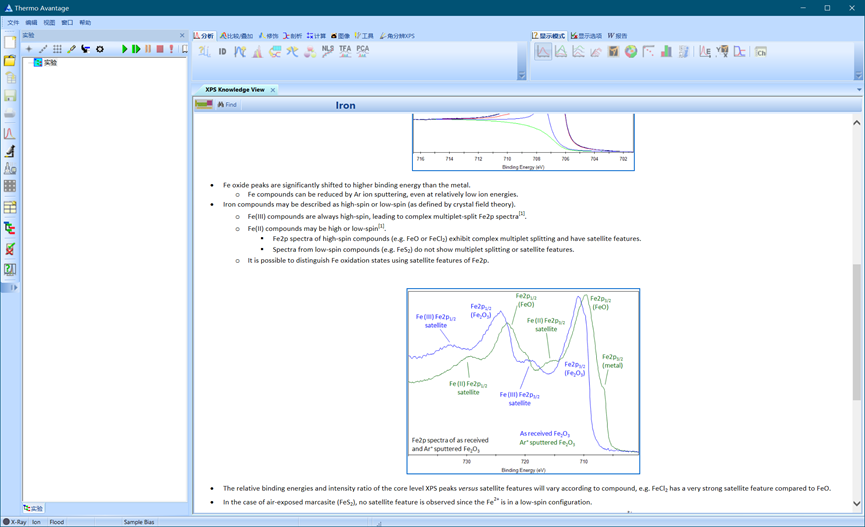
图4.18 Avantage数据库中关于Fe 2p卫星峰的分析方法
4.3.2 Mn 3s
由于不同化合价Mn的Mn 2p轨道结合能差距很小,难以分析出具体的化合价。
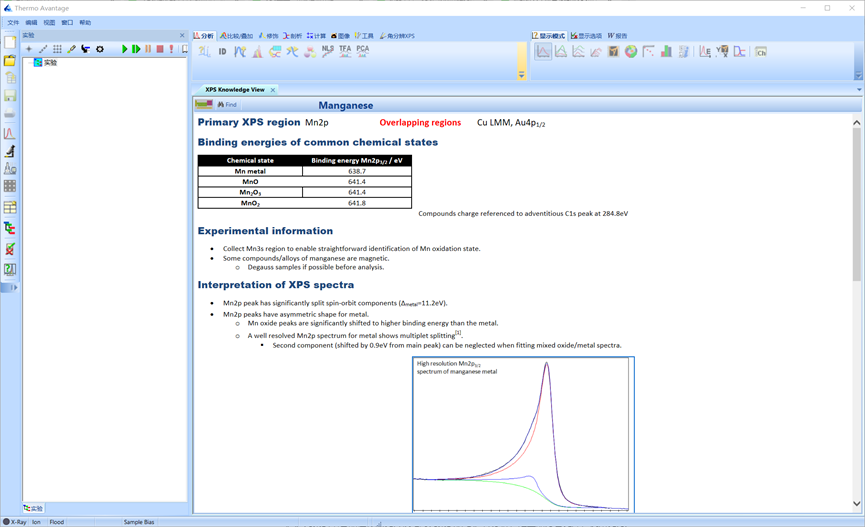
图4.19 Mn 2p轨道结合能差距小,难以区分
因此可以使用Mn 3s轨道上两个峰的结合能差对Mn的化合价进行分析。
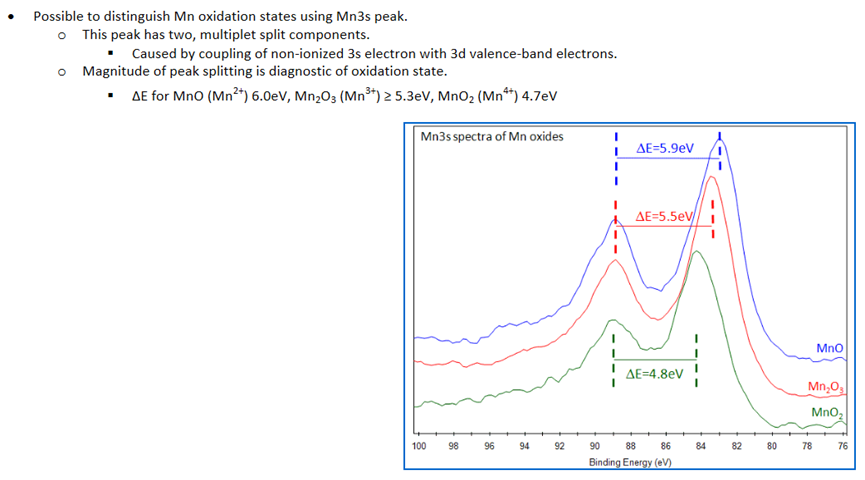
图4.20 通过Mn 3s轨道分析Mn的化合价
4.3.3 S 2p
p轨道为自旋分裂峰,但是结合能差很小,只有约1.16 eV,在图谱中难以分辨出所有峰型,因此部分文献在分峰中不严谨地按照单峰拟合的方法进行分析。严谨的分峰拟合应添加双峰,因此S 2p的精细谱一定会被分成偶数个峰,且每对峰均符合结合能差和峰面积比例。
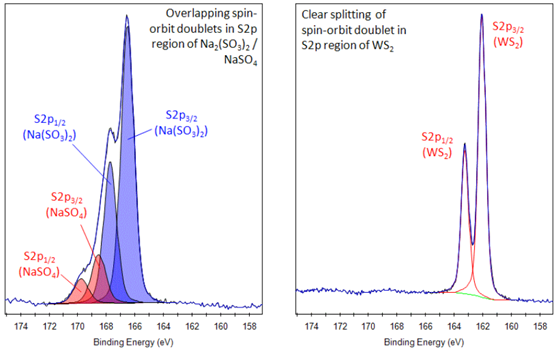
图4.21 不同材料中S 2p精细谱的分峰拟合
总之,每种元素的XPS精细谱都有自己独特的分析方法,本文难以一一列举,在实际测试前后还需要同学们调研文献和查阅数据库,以便尽可能准确地实现对材料的XPS采谱和分析。
5. XPS在能源材料领域的前沿应用
5.1 原位电化学XPS
与传统的在超真空条件下工作的XPS技术不同,近常压光电子能谱(NAP-XPS)可以研究催化剂颗粒在较高温度下、一定压力(1 Torr左右)气体环境中的表面化学。近十年来,NAP-XPS广泛用于研究多种催化剂在反应条件下的表面化学,包括单晶模型催化剂、金属颗粒催化剂、金属氧化物与碳化物催化剂。NAP-XPS的发展也推动了原位XPS的进步。2021发表在Nature的工作报道了孤立Pt单原子(Pt1)及亚纳米的Pt团簇(Ptn)共同负载α相碳化钼(α-MoC)催化剂(Pt1–Ptn/α-MoC),其能在313 K的低温下催化水煤气转换反应。如图5.1所示,催化剂在活化后显示出C和Mo的特征峰,分别位于284.8和230 eV。从O 1s XPS谱可以观察到位于530.3 eV的α-MoC表面残余O信号峰(O r)。当在处于室温下的XPS样品室引入5 mbar的气态H2O,除了能原位观察到位于534.8 eV的气相H=O信号峰(H2O g)以及O r,还新发现位于532.3 eV的信号峰,其可归属于α-MoC表面的吸附OH物种(OH ads)。此外,使用瞬态动力学分析(TKA)证实了H2的产生。这些结果证明2 wt%(Pt1–Ptn)/α-MoC在室温下可以解离H2O,产生H2以及吸附在α-MoC表面的OH物种。当表面OH物种形成后,气相H2O被赶出并在不同温度下引入CO。当在303 K温度下引入5 mabr CO,出现位于537.6 eV的气相CO(CO g)以及533.8 eV的吸附CO峰(CO ads)。表面OH物种的信号峰强度从323 K开始显著降低。但是,即使反应温度提升到423 K,在α-MoC表面仍然能观察到很强的OH物种的信号峰,证明在α-MoC表面存在两种羟基物种:一种靠近可吸附CO的Pt,另一种是远离Pt的羟基物种。靠近Pt的羟基物种可以被吸附的CO消耗,而远离Pt的羟基物种不会参与WGS反应。不参与反应的羟基物种可导致催化剂载体氧化,因而使催化剂失活。
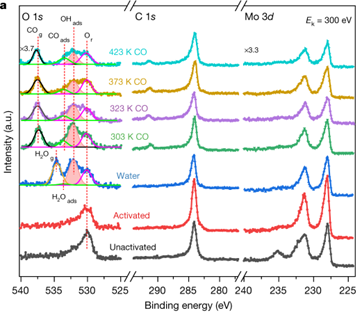
图5.1 (Pt1–Ptn)/α-MoC的催化剂在不同条件下的NAP-XPS谱
作者紧接着使用原位XPS评估0.02 wt% Pt1/α-MoC和2 wt% (Pt1–Ptn)/α-MoC催化剂的稳定性。原位XPS结果显示0.02 wt% Pt1/α-MoC催化剂的Mo 3d谱明显向高价态移动,表明0.02 wt% Pt1/α-MoC在反应后载体发生氧化,解释了其催化活性显著下降的原因。相反,2 wt% (Pt1–Ptn)/α-MoC的Mo 3d谱在反应前后没有发生位置移动,对应其良好的催化稳定性(图5.2)。
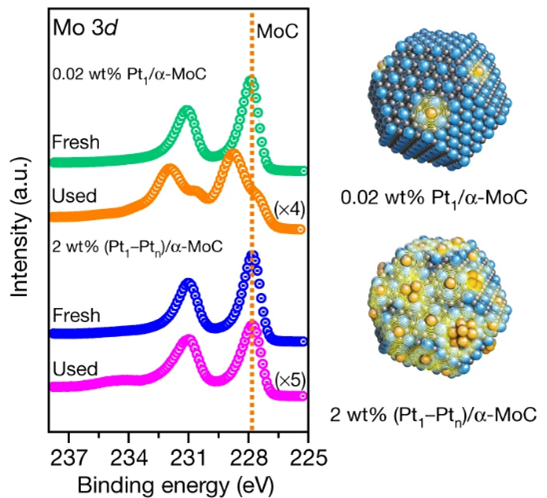
图5.2 0.02 wt% Pt1/α-MoC 和2 wt%(Pt1–Ptn)/α-MoC催化剂在反应前后Mo 3d的原位XPS谱
原位XPS作为一种新型表征手段,可以用来对表面反应进行表征和观测,还可以用于SEI生成机理的研究,是科研中的一大助力。
5.2 XPS深度分析
XPS深度分析是通过Ar离子枪对样品表面进行溅射剥离,控制合适的溅射强度及溅射时间,将样品表面刻蚀到一定深度,然后进行取谱分析。2022年发表在eScience上的一篇文章就用到了XPS深度分析。这篇文章提出了伪浓缩电解质的概念,伪浓缩电解质可以形成坚固的富含LiF的固态电解质界面(SEI)。首先使用XPS对负极进行表征,发现在负极表面,C-F信号在空白电解质中最高。在三种不同电解质BA、PD和4-PBA里形成的SEI中,Li-F是主要成分,还发现了少量化合物(如Li2O/Li-B-O和Li2CO3)。根据XPS的F 1s谱,发现BA、PD和4-PBA电解质中的高LiF含量可能是Li||Li电池优异的长期稳定性的原因。BA电解质中LiF含量最高,但这并没有产生最佳性能。进一步使用XPS深度分析发现,空白电解质和BA电解质的SEI中存在不均匀、脆弱的镶嵌结构。相比之下,4-PBA电解质中形成的SEI显示出双层结构,包括外部有机层(Or.C)和内部无机(LiF)层,这种结构更加稳定。
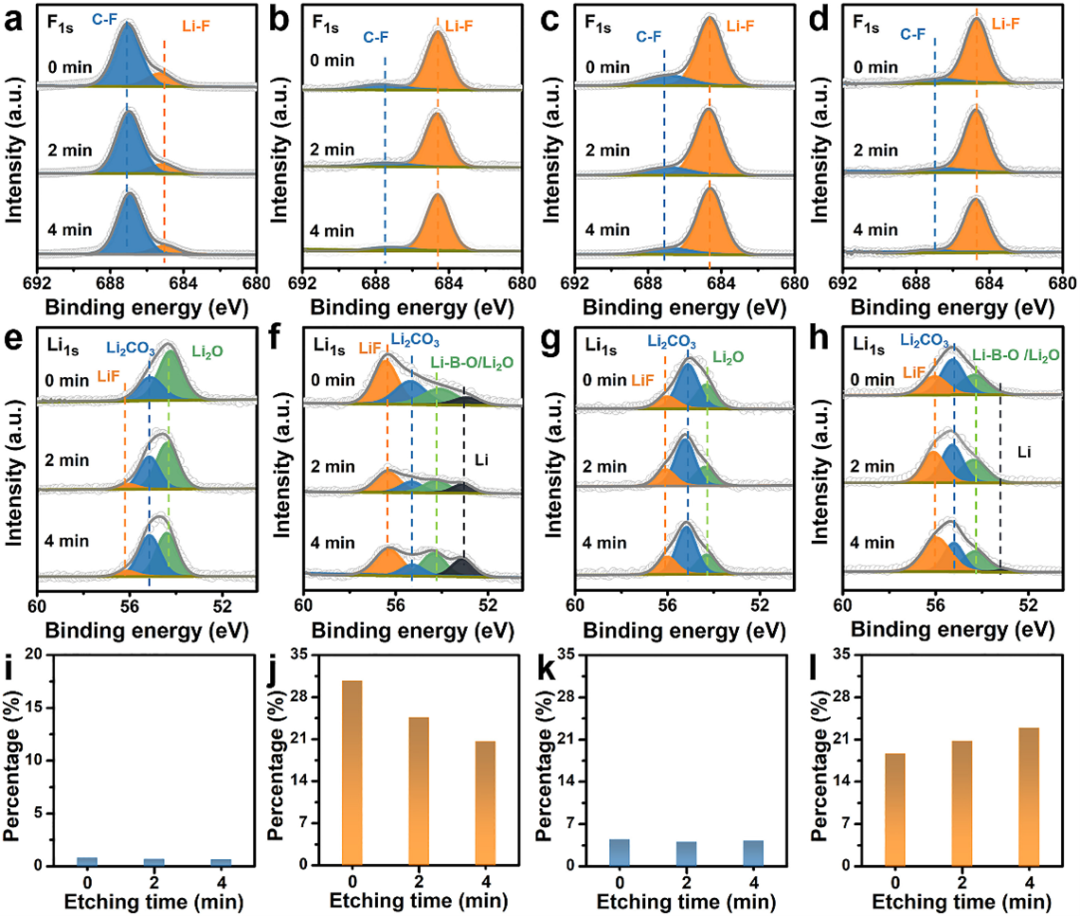
图5.3 使用XPS深度分析对SEI不同深度的性质进行表征。Li‖Li对称电池中的(a–d) F 1s和 (e–h) Li 1s 在使用 (a, e) 空白、(b, f) 1.0 wt% BA,(c,g)1.0 wt% PD,和(d,h)1.0 wt% 4-PBA 电解质进行10次循环测试后的XPS谱图。从(i) 空白、(j) 1.0 wt% BA、(k) 1.0 wt% PD 和 (l) 1.0 wt% 4-PBA 电解质的 F 1s 光谱中量化LiF含量
XPS深度分析在二维结构和材料表面研究方面十分重要,可以用来直观的分析微区内不同厚度的元素含量和化合状态。
6. XPS的局限性
XPS作为一种灵敏度很高的表面分析方法,实验结果难免会受到各种因素的影响,比如表面形貌、荷电效应和离子溅射效应等,因此在构筑界面和测试过程中,特别是对于原位实验,需要考虑如何减少干扰因素的影响。
XPS目前不能用于液体和气体的分析,对样品的表面要求非常高,需要样品表面干净、平整,并且不能有氧化层或其他污染物。
XPS分析需要在高真空环境下进行,一些敏感和生物样品可能难以进行测试。
XPS分析的深度有限,只能分析样品表面几纳米的深度范围。因此,对于一些厚度较大的样品或者需要分析样品内部结构的情况,XPS可能不适用。
XPS是一种微区测试,而且探测深度极浅,因此难以反应整个样品的情况。
结语
尽管XPS的测试流程是标准化的,但是在分析方面带有很强的主观因素,分析的准确与否取决于对这项技术的了解程度。由于对XPS分析方法的不熟悉,现在很多已发表的文献中对XPS分析的准确性还有待商榷。文献中XPS分析常见的问题包括去基线错误、元素价态标注错误、自旋分裂峰峰强比例错误、峰谱重叠、卫星峰误标、p、d、f轨道未按照分裂峰进行分峰等等。在分析XPS之前一定要全面地检索数据库及相关文献,尽可能准确地对XPS谱峰进行分峰及标定。
随着XPS技术的不断发展和优化,其在各个领域的应用将更加广泛,而与其他表面分析技术(如UPS﹑AES﹑SIMS﹑STM等)联合应用,也会使分析结果更全面﹑完善﹑正确﹑可靠。
参考书籍
[1] 吴正龙. 表面分析 XPS和AES 引论[M]. 上海:华东理工大学出版社, 2008.01.
[2] 宋廷鲁,邹美帅,鲁德凤. X射线光电子能谱数据分析[M]. 北京:北京理工大学出版社, 2022.11.
[3] 刘世宏. X射线光电子能谱分析[M]. 北京:科学出版社, 1988.10.
[4] D.Briggs. X射线与紫外光电子能谱[M]. 北京:北京大学出版社, 1984.02.
参考文献
[1] J. Zhang, L. Zhang, W. Wang, et al. In Situ Irradiated X-ray Photoelectron Spectroscopy Investigation on Electron Transfer Mechanism in S-Scheme Photocatalyst[J]. The Journal of Physical Chemistry Letters, 2022, 13, 8462.
[2] Y Xu, C Zhang, M Zhou, et al. Highly nitrogen doped carbon nanofibers with superior rate capability and cyclability for potassium ion batteries[J]. Nature Communications, 2018, 9, 1720.
[3] J Liu, S Zou, L Xiao, et al. Well-dispersed bimetallic nanoparticles confined in mesoporous metal oxides and their optimized catalytic activity for nitrobenzene hydrogenation[J]. Catalysis Science & Technology, 2014, 4, 441-446.
[4] J Chastain. Handbook of X-ray photoelectron spectroscopy[J]. Perkin-Elmer Corporation, 1992, 40, 221.
[5] M C Biesinger, B P Payne, A P Grosvenor, et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni[J]. Applied Surface Science, 2011, 257, 2717-2730.
[6] X Zhang, M Zhang, Y Deng, et al. A stable low-temperature H2-production catalyst by crowding Pt on α-MoC[J]. Nature, 2021, 589, 396-401.
[7] H Wang, J Liu, J He, et al. Pseudo-concentrated electrolytes for lithium metal batteries[J]. eScience, 2022, 2, 557-565.
本文源自微信公众号:eScience期刊
原文标题:《【eScience-干货】一文学会XPS测试原理与数据处理》
原文链接:https://mp.weixin.qq.com/s/zMZV9R0qBUUi115dvSDRDg
本转载仅出于分享优质测试干货,旨在传递更多观点,并不代表赞同其全部观点或证实其内容的真实性。文章中所包含的图片、音频、视频等素材的版权均归原作者所有。如有侵权请告知删除。