

什么是MXene
近年来,一个新兴的二维材料家族在科研界频频出现,它就是MXene。MXene通常由碳、氮与过渡金属原子组成,层状结构像是一片“金属三明治”,表面常常带有-OH、-O、-F等官能团修饰。下图系统分类了MXene材料,包括单M元素(如Ti2C)、固溶体M元素(如(Ti0.5Nb0.5)2C)及有序双M元素(如Mo2TiC2)三大类,标注了实验/理论状态及化学式。
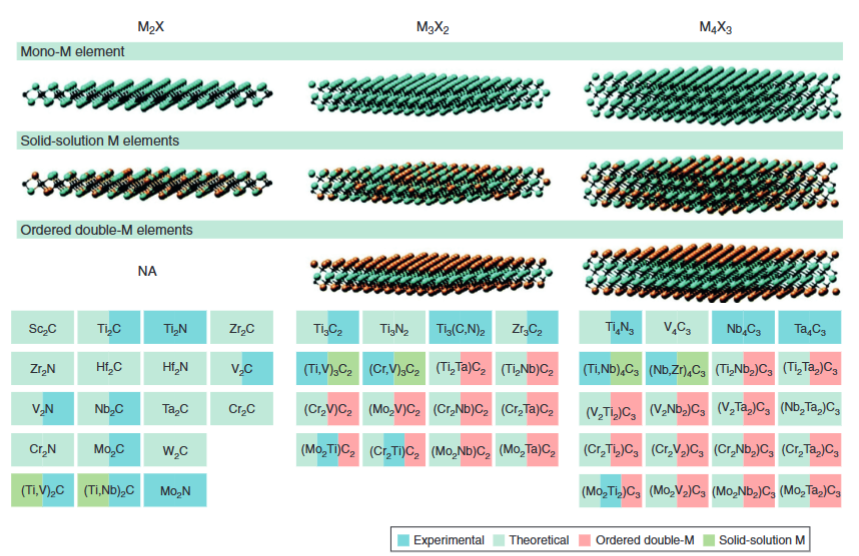

DOI:10.1515/nanoph-2019-0571
由于这种独特结构,MXene既具备金属般的高导电性,又有良好的化学可调性,被称为“多面手”材料。那么,MXene究竟能做什么?从应用层面看,它可以用于电池电极、超级电容器、催化剂、传感器甚至海水淡化膜。但如果换个角度,从计算化学入手,我们就会发现MXene其实是一个非常适合做理论预测和模拟研究的材料“试验田”。
如何理解MXene
计算化学能帮助我们在原子层面揭开MXene的秘密。最常用的方法是密度泛函理论(DFT),它可以告诉我们MXene的能带结构、电子密度分布以及表面官能团对性能的影响。比如,我们能通过计算得知某种表面修饰会让MXene变得更亲水,或者更适合吸附离子。进一步的计算还可以预测离子在MXene表面的吸附能,判断它是否适合作为电池电极材料。
下图展示了使用DFT来研究插入金属的选择显著影响MXene性能,特别是导电性和电荷存储行为。MXene层内的Ti原子被氧化到不同程度,取决于插入的过渡金属,改变费米能级附近的电子态密度。
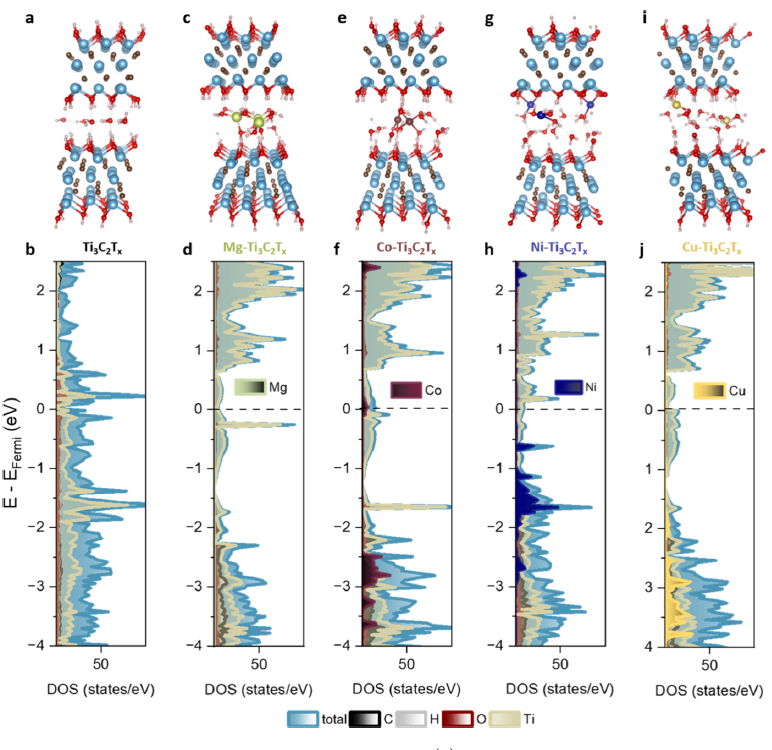
DOI:10.1021/acsnano.5c06170
如果把视野扩大一些,还可以用分子动力学(MD)模拟 来研究MXene在溶液中的稳定性,以及水分子、离子如何穿梭在MXene片层之间。这些结果不仅帮助解释实验现象,还能提前筛选出更有前景的材料组合,从而节省大量时间和实验成本。可以说,计算化学既能看清MXene中分子移动细节,也能预测MXene的未来优化方案。
下图详细展示了使用AIMD来研究水分子在MXene层间的穿梭过程:包括H+、H2O和Zn的MSD曲线,显示扩散速率差异,以及时间帧序列图,演示水分子在不同条件下(如纯水、Zn插入、H插入)如何在层间移动、吸附和解吸。
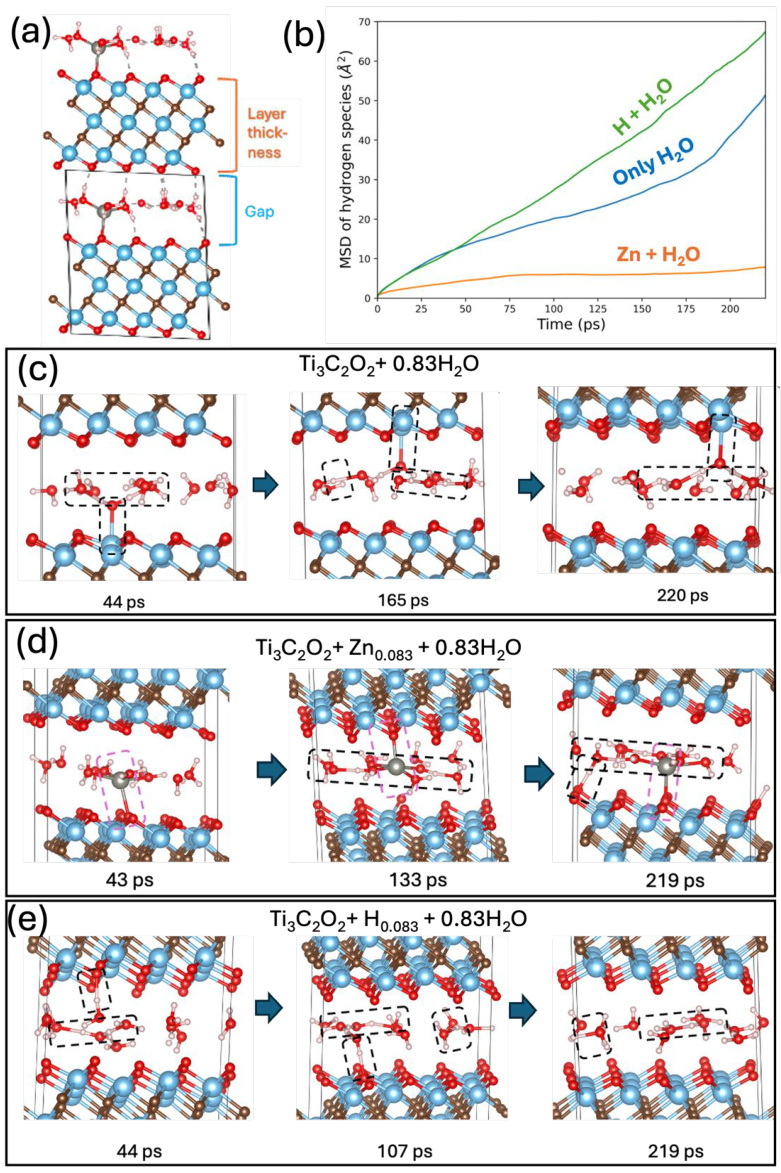
DOI: 10.1021/acsami.5c06056
研究MXene的意义
为什么我们要对MXene做这么多计算?原因在于它的潜力太大,而可能性也太多。MXene的化学组成都能灵活调节,不同的金属、碳氮比例以及表面官能团都会带来截然不同的性能。如果只靠实验逐一尝试,耗费巨大且效率低下。而通过计算,我们可以快速绘制出性能地图,比如哪种MXene在锂离子电池中最稳定,哪种在催化分解水产氢时最活跃,哪种在屏蔽电磁波方面效果最好。
计算不仅能帮我们找到答案,还能揭示背后的机理,为新设计提供指导。未来,随着计算与机器学习、大数据方法结合,MXene研究将更加高效,甚至可能实现“按需设计”材料。对于初学者来说,MXene就是一个展示计算化学如何服务于新材料发展的最佳例子:从微观结构出发,通过模拟与预测,加速推动实验和应用的落地。
总结
MXene因其独特的二维层状结构和可调节的表面化学性质,被广泛视为未来材料科学的重要候选。它不仅在能源存储、催化和传感器等领域展现巨大潜力,更因为能通过计算化学进行深入研究而显得格外特别。DFT可以帮助我们理解其电子性质和反应活性,分子动力学能揭示其在复杂环境中的动态行为,而更大尺度的模拟则能预测实际应用中的稳定性与性能。
对于科研人员而言,计算研究不仅是解释实验现象的工具,更是提前筛选材料、加快研发进程的利器。未来,随着计算方法与机器学习结合,MXene研究将更加高效和系统,甚至可能实现材料的按需设计。对于初学者来说,MXene是一个理解“计算如何驱动新材料发展”的理想案例。