

标准的分子动力学模拟通过数值求解牛顿运动方程,来追踪系统中每个原子的运动轨迹,从而预测材料的宏观性质。而外磁场下的分子动力学模拟,则是在此基础上引入了磁场与体系中粒子间的相互作用,使得我们能够观察和研究系统在磁场驱动下的动态演化过程。
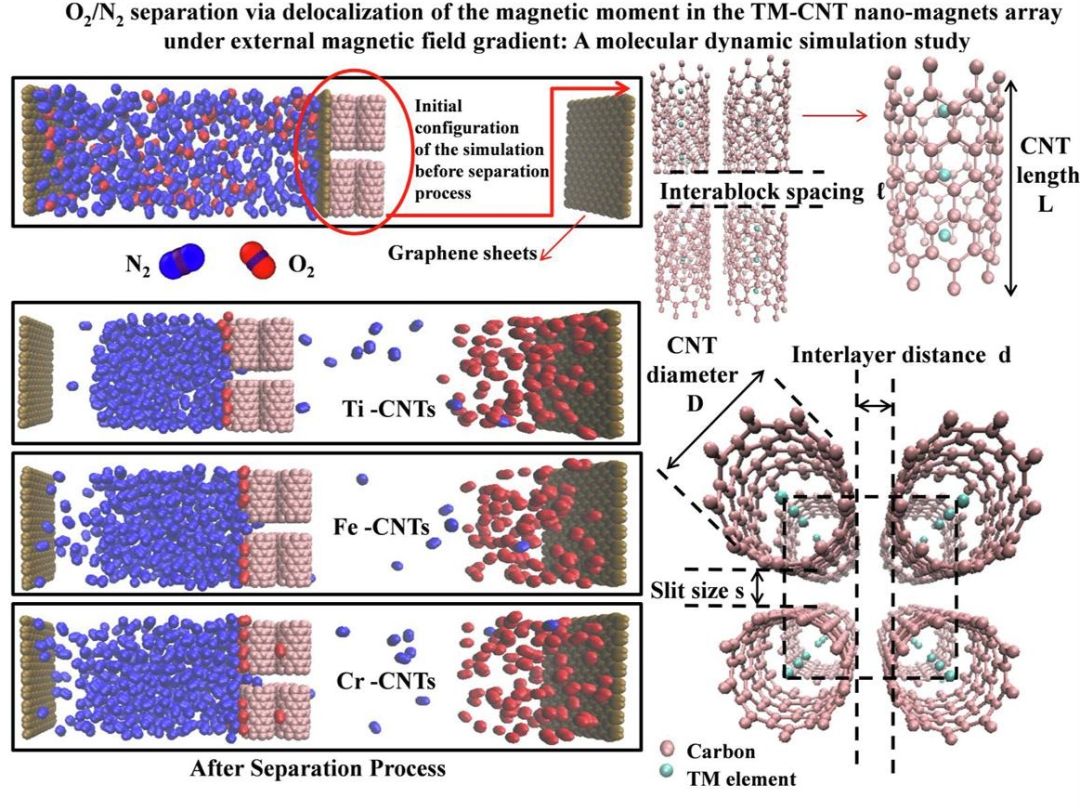

DOI: 10.1016/j.apsusc.2024.161706
其理论核心是在经典力场的基础上,额外增加描述磁场相互作用的力或能量项。根据体系内粒子的物理属性,磁场的引入主要通过两种关键的物理机制:洛伦兹力(Lorentz Force) 和自旋–磁场耦合(Spin-Magnetic Field Coupling)。
洛伦兹力:对于体系中的带电粒子(如离子),其在磁场B 中以速度 v 运动时,会受到洛伦兹力的作用。这个力的大小和方向由以下公式决定:
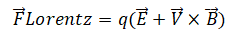
其中,q是粒子的电荷,E是电场强度。在仅考虑磁场的情况下,洛伦兹力始终垂直于粒子的速度方向,因此它不对粒子做功,只改变其运动方向,导致带电粒子在垂直于磁场的平面内做回旋运动。
自旋–磁场耦合:对于具有固有磁矩(即“自旋”,S)的粒子,如磁性纳米颗粒或某些原子,外磁场 B 会对其施加一个力矩(Torque),这种相互作用通常由塞曼(Zeeman)能量项描述。该力矩不直接产生平动,而是驱动磁矩绕磁场方向进动。这一过程通常由朗道–里夫希茨–吉尔伯特(Landau-Lifshitz-Gilbert, LLG)方程描述:

其中,γ是旋磁比,α是阻尼系数,Beff是包含外场、各向异性场和交换作用的有效磁场。在很多模拟中,原子或粒子的平动(通过牛顿方程)和其磁矩的转动(通过LLG方程)被耦合在一起,这种方法被称为“自旋–晶格动力学”(Spin-Lattice Dynamics)模拟。
因此,外磁场下的MD模拟并非单一模型,而是一个集成了多种物理图像的混合模拟框架。它将粒子的空间运动与电荷、磁矩等内禀属性紧密结合,从而能更真实地反映物理世界的复杂性。
系统准备与参数设定
与常规MD模拟类似,第一步是构建初始模型。这包括定义模拟盒子、填充原子或分子、并选择合适的力场来描述粒子间的常规相互作用(如Lennard-Jones势、库仑势等)。关键的不同之处在于,研究者需要为体系中的粒子赋予额外的磁学属性:
对于带电体系:为相关原子或离子设定电荷。
对于磁性体系:为每个磁性粒子或原子定义其磁矩大小和初始方向。
同时,需要明确定义外部磁场的参数,包括磁感应强度B的大小、方向,以及其是否随时间变化(静态场或动态场)。
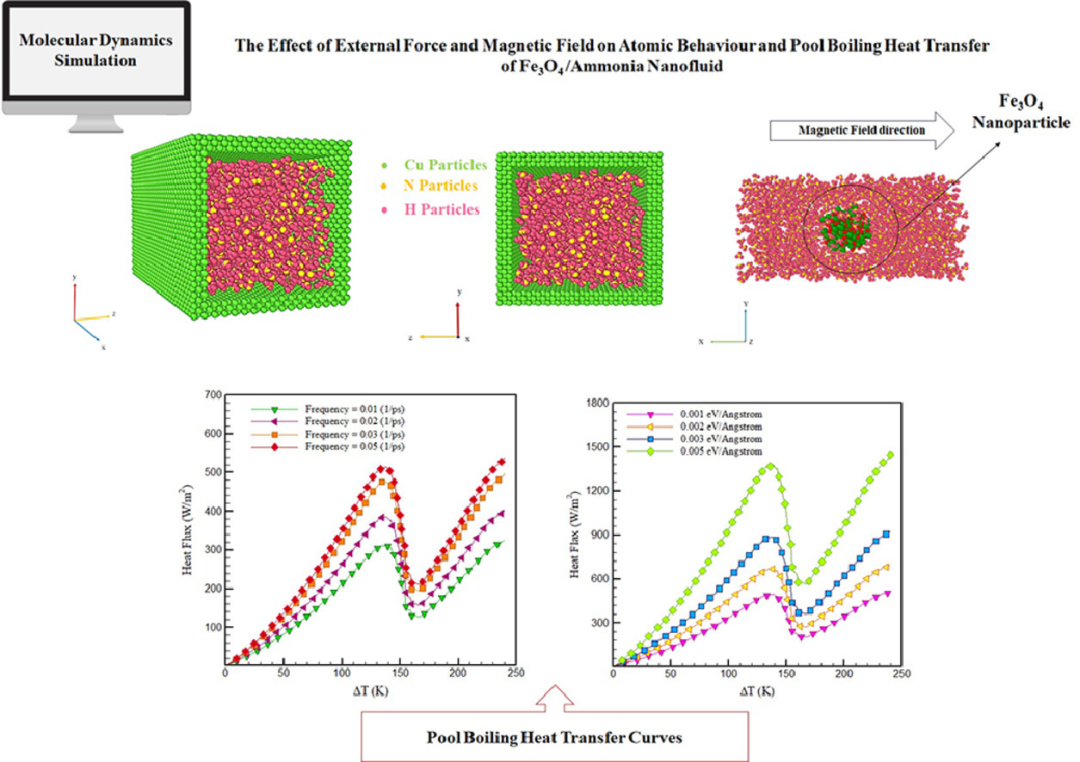
DOI: 10.1016/j.jtice.2023.104781
模拟软件与磁场实现
目前,主流的开源MD模拟软件包(如LAMMPS, GROMACS, AMBER等)拥有强大的可扩展性。虽然它们可能没有直接内置“一键式”的磁场模拟功能,但通过其灵活的框架,实现磁场效应是完全可能的。
运动方程的积分算法
这是技术实现上的核心难点。标准的积分算法(如Velocity-Verlet)在处理保守力时表现优异且能保证能量守恒,但洛伦兹力是速度依赖的非保守力,直接应用标准算法会导致轨迹不稳定和能量漂移。
为此,学术界发展了专门的算法,其中最著名和最常用的是鲍里斯算法(Boris Algorithm)。该算法的核心思想是将一个时间步内的运动分解为三步:
1、半个时间步的电场或常规力导致的“加速”(Kick)。
2、一个完整的磁场导致的“旋转”(Rotation),这一步不改变速度大小,解析地求解了粒子在磁场中的旋转运动。
3、另半个时间步的电场或常规力导致的“加速”(Kick)。
这种“Kick-Drift-Kick”的变体形式巧妙地处理了洛伦兹力,能在较大时间步长下依然保持极高的精度和长期稳定性,是等离子体物理和带电粒子模拟领域的金标准。
模拟执行与结果分析
与常规MD一样,模拟包含能量最小化、系统弛豫(平衡)和生产(数据采集)等阶段。在分析阶段,除了常规的径向分布函数、温度、压力等参数外,研究者更关注磁场特有的输出:
粒子轨迹:观察带电粒子是否呈现螺旋运动。
磁矩取向:分析磁性粒子的磁矩是否沿外场方向排列,计算序参量。
宏观磁化强度:通过统计体系中所有磁矩的矢量和,可以绘制磁化曲线(M-H curve),即宏观磁化强度随外场强度的变化关系,并与实验数据对比。
微观结构:对于磁流变液或磁性纳米颗粒体系,可视化分析粒子是否形成链状、柱状等特定结构。
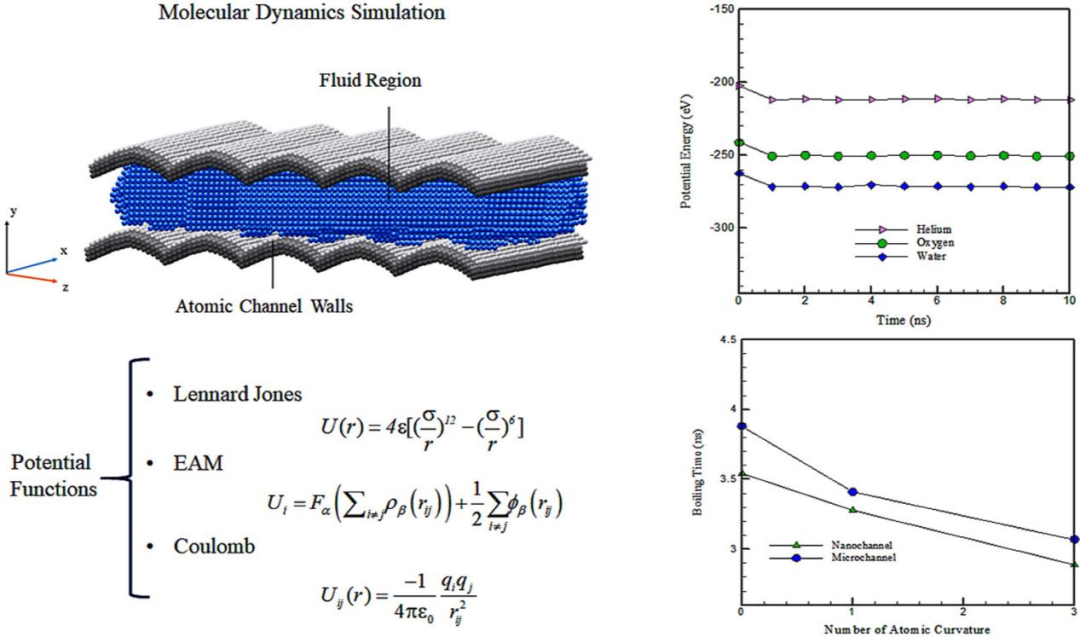
DOI: 10.1016/j.csite.2022.102105
尽管在软件实现和算法选择上存在一定的技术挑战,但随着计算能力的提升和理论方法的发展,该技术正在成为推动磁性功能材料、生物医学技术和自旋电子学等领域创新的重要引擎。未来,发展更高效、更精确的多物理场耦合模拟方法,将是该领域持续前进的关键方向。