

什么是COF材料
COF全称是共价有机框架(Covalent Organic Framework),可以把它想像成用有机分子像积木一样搭成的三维或二维“积木房子”。这些“积木”之间通过共价键牢牢连接,形成高度有序、孔洞可控的网状结构。下图展示了具有分层堆叠结构的二维COF。

DOI:10.1021/acs.chemrev.9b00550
对普通读者来说,重要的点有三条:一是全由轻元素(C、H、O、N等)构成,二是框架内有规则的孔隙,三是结构可通过选择不同的有机单元来设计。
因此COF看起来像是“有孔的有机晶体”,既有固体的稳定框架,又有多孔材料的大表面积,适合做分子吸附、催化或储能的“微型工厂”。
如何计算COF
计算化学就像给COF装上显微镜和“预见未来”的工具。通过量子化学计算(比如密度泛函理论,DFT)可以预测单层或晶胞级别的电子结构、带隙、吸附能等。
下图通过DFT计算展示了COFs的强亲锂性和高富氮结构具有多个吸附位点和高的Li吸附能,自发形成具有丰富Li-N和高度有序孔结构的刚性有机/无机杂化保护层,从而诱导均匀的Li+通量和Li镀/剥离,降低了Li+迁移能垒,增强了Li+的迁移率,抑制了锂枝晶的生长。
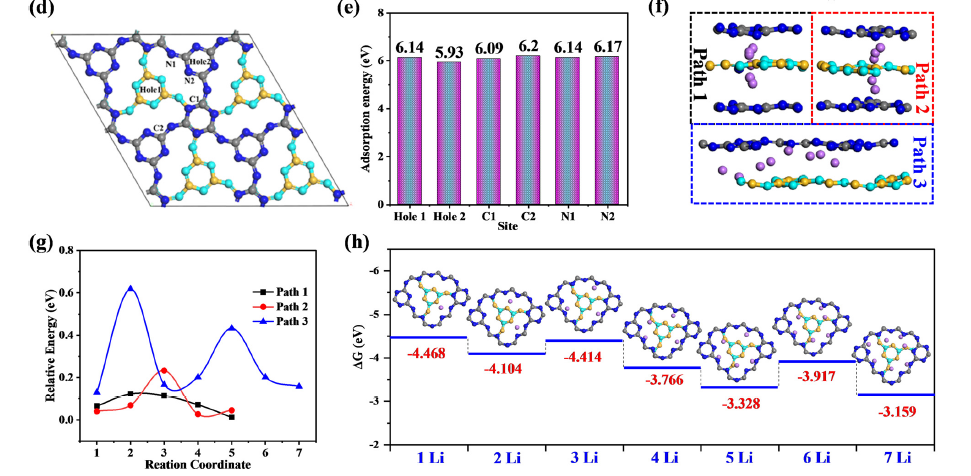
DOI: 10.1039/D3EE02803H
通过分子动力学(MD)可以模拟在不同温度、溶剂或电场下分子在孔内的运动和扩散行为;通过力场和蒙特卡洛方法可以研究气体在孔道内的吸附等温线。
例如下图展示了通过MD模拟揭示了离子跨COF通道传输的微观机制,首次观察到Mg2+在与通道中的磺酸基团相互作用时会发生部分脱水,这一发现为理解选择性传输机制提供了关键证据。
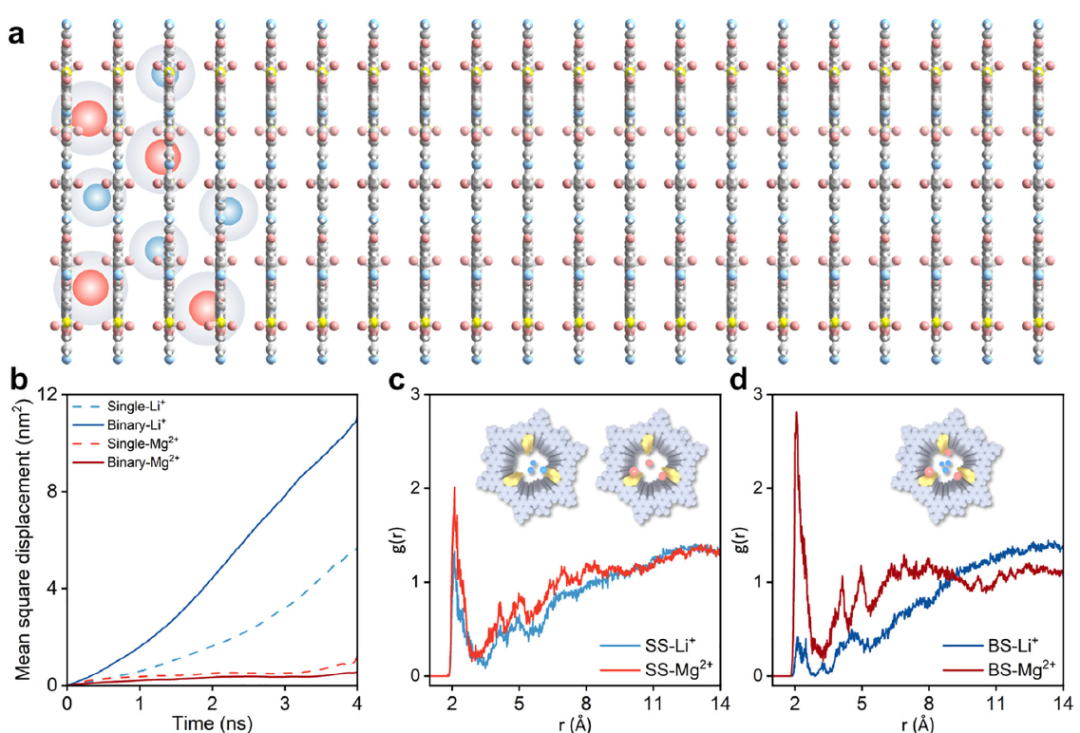
DOI: 10.1002/adma.202414898
简单说,计算化学帮助我们从原子和电子层面理解COF为什么会有某种物理化学性质—比如为什么某个孔径大小更利于甲烷吸附,或者为什么某种连接方式会让电子更容易移动。
对初学者而言,想象把一个看不见的分子世界用“方程”和“模拟”变成可视化的能量曲线、轨道图和运动轨迹,就很直观了。
计算化学不是单纯“算数”,而是把原子级别的假设变成可验证的预测。对于COF这样高度可设计的材料,实验上合成一个新骨架既耗时又昂贵。
计算可以先在“虚拟试验台”上筛选单体组合、预测稳定性与性能,帮研究者把有限的实验资源集中在最有希望的候选上。
更重要的是,计算能直接揭示难以用实验直接观测的微观机制——比如电子如何沿着共轭链传导、某个位点为何更容易与气体分子结合、或者缺陷如何改变离子扩散路径。
COF的功能
COF的功能非常多样,常见的包括气体储存与分离(比如储氢、吸附二氧化碳、分离甲烷/二氧化碳等)、催化(把活性位点固定在孔内以提高反应速率和选择性)、吸附/去除污染物(有机分子、重金属离子)、传感(孔内与目标分子相互作用导致光学或电学信号变化)、以及能量存储与传输(用于超级电容、电池电极或导电膜)、药物递送与缓释、以及光电/电子器件(光吸收、发光、载流子传输)。
从计算化学角度看,这些功能可以用具体可量化的指标来描述和优化:
1、计算吸附能和吸附等温线预测哪种分子会被优先吸附;
2、通过分子动力学算出孔内扩散系数,判断运输快慢;
3、用过渡态搜索和反应能垒计算催化活性;
4、用电子结构计算(如带隙、轨道分布)评估电导和光学性能;
5、还可以模拟热力学和力学稳定性,判断在实际环境下是否耐用。
总之,计算给我们提供了“先验量化”的能力,能在动手合成之前就筛出最有希望完成特定功能的COF。
总结
COF的核心优势在于“分子可编程”:通过选择节点与连接体、调控官能团与拓扑,即可在原子尺度上把孔径、极性、共轭程度与化学环境精准写入框架。
与传统多孔材料相比,COF兼具结构有序与化学可塑性,非常适合承载特定分子识别与传输任务。
计算化学让这种“可编程”从理念变成工程化流程:
首先用力场/DFT完成晶胞几何优化与稳定性评估,得到可靠的孔结构与能量基线;
随后以DFT计算关键吸附物(如CO2、H2O、金属离子)在不同位点的结合能与电荷转移,识别真正的活性位点;
再以GCMC生成多温压下的吸附等温线与选择性曲线,预判混合气体或多组分水溶液中的竞争吸附;
对传输与膜分离问题,最后用MD获取自扩散系数、停留时间分布与框架柔性对瞬时孔径的影响。
最后通过“计算—实验—再计算”的迭代,COF在气体分离、海水净化、环境治理、储能电极与催化等方向的开发效率与成功率显著提升。