本文详细介绍了共价有机框架(COF)的基本组成、结构类型及其在气体吸附和催化性能研究中的应用。COF通过共价键连接有机分子,形成规则的孔道结构和周期性排列,具有优异的吸附和催化性能。
在气体吸附方面,通过分子动力学模拟揭示了COF对H₂和CO₂的选择性分离机制;在催化性能研究中,DFT计算阐明了硫修饰对COF光催化活性的优化作用。这些研究为COF材料的设计与应用提供了重要的理论依据和实验指导。
COF 结构的基本组成与类型
COF结构由有机分子通过共价键连接而成,构成要素包括有机连接体和连接方式。有机连接体通常为具有特定几何形状和反应活性的有机分子,如含硼、碳、氮、氧等元素的化合物,它们决定了COF的基本框架和化学性质。连接方式则有多种,如B-O、C-N、B-N 等共价键,这些连接方式赋予COF高度的稳定性和可控性。

根据结构维度和拓扑结构,COF可分为不同类型。从维度上,有二维(2D)COF和三维(3D)COF。2DCOF通常具有层状结构,层内通过共价键连接形成规整的孔道结构,层间则通过较弱的相互作用(如范德华力)堆叠。3DCOF则形成三维贯通的网络结构,具有更复杂的孔道体系和更高的比表面积。
在拓扑结构方面,COF存在多种形式,如蜂窝状、四方晶格、立方晶格等拓扑结构,不同拓扑结构决定了COF的孔道形状、尺寸和排列方式,进而影响其物理化学性质。
COF具有规则的晶体结构和周期性排列特点。理论计算表明,其晶体结构中的原子按特定的晶格常数和空间群规则排列,形成周期性的结构单元。这种周期性使得COF在宏观上表现出均匀的物理化学性质,如在X射线衍射图谱中呈现出特征性的衍射峰,对应于晶体结构的不同晶面间距。
晶体结构的周期性对COF性能有重要影响。一方面,周期性结构为分子吸附提供了均匀的吸附位点,有利于实现对特定分子的选择性吸附。例如,在气体吸附应用中,COF的周期性孔道结构可与特定气体分子的尺寸和形状相匹配,通过分子与孔壁的相互作用实现高效吸附和分离。
另一方面,周期性结构影响COF的电子传输性质,规则排列的原子轨道有利于电子的离域和传导,对COF在光电领域的应用具有重要意义,如影响其作为半导体材料的载流子迁移率和光电转换效率。
COF的孔道结构丰富多样,具有明确的孔道形状和尺寸分布。理论计算可精确预测COF的孔道结构参数,如孔径大小、孔壁粗糙度和孔道连通性。常见的COF孔道形状有圆形、六边形、四边形等,孔径范围涵盖微孔(小于2nm)到介孔(2-50nm)。
孔道结构和尺寸对分子吸附和扩散有显著影响。较小的微孔尺寸适合吸附小分子,如氢气、二氧化碳等,通过分子与孔壁的强相互作用实现高吸附容量。
较大的介孔则有利于大分子的扩散和吸附,如在催化反应中,为反应物和产物分子提供足够的扩散空间,提高反应速率。此外,孔道的连通性决定了分子在COF内部的扩散路径,高连通性的孔道结构能促进分子快速传输,提高材料的吸附和分离效率。
通过理论计算研究COF的电子结构,可获得其能带结构、电子云分布和电荷转移等信息。COF的电子结构决定了其电学、光学和催化等性能。例如,具有合适能带结构的COF可表现出半导体特性,能用于光电器件;电子云分布和电荷转移情况影响COF与吸附分子之间的相互作用,进而影响其吸附和催化活性。
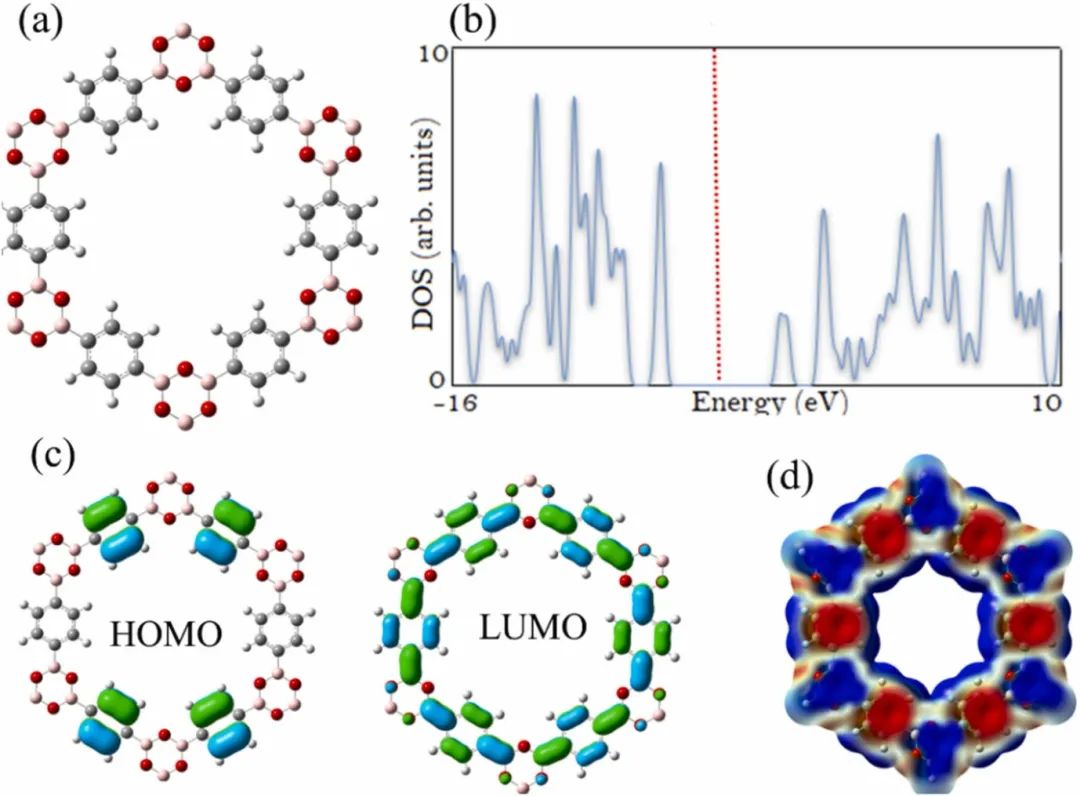
COF中的化学键特性对其结构稳定性和化学性质至关重要。不同类型的共价键(如C-N、B-O等)具有不同的键能和化学活性,决定了COF在不同化学环境下的稳定性。
较强的共价键赋予COF良好的热稳定性和化学稳定性,使其能在高温、酸碱等苛刻条件下保持结构完整性;而化学键的化学活性则决定了COF的化学反应活性,可通过选择不同的连接体和连接方式来调控COF的化学性质,实现对特定化学反应的催化或参与。
COF在气体吸附研究中的应用
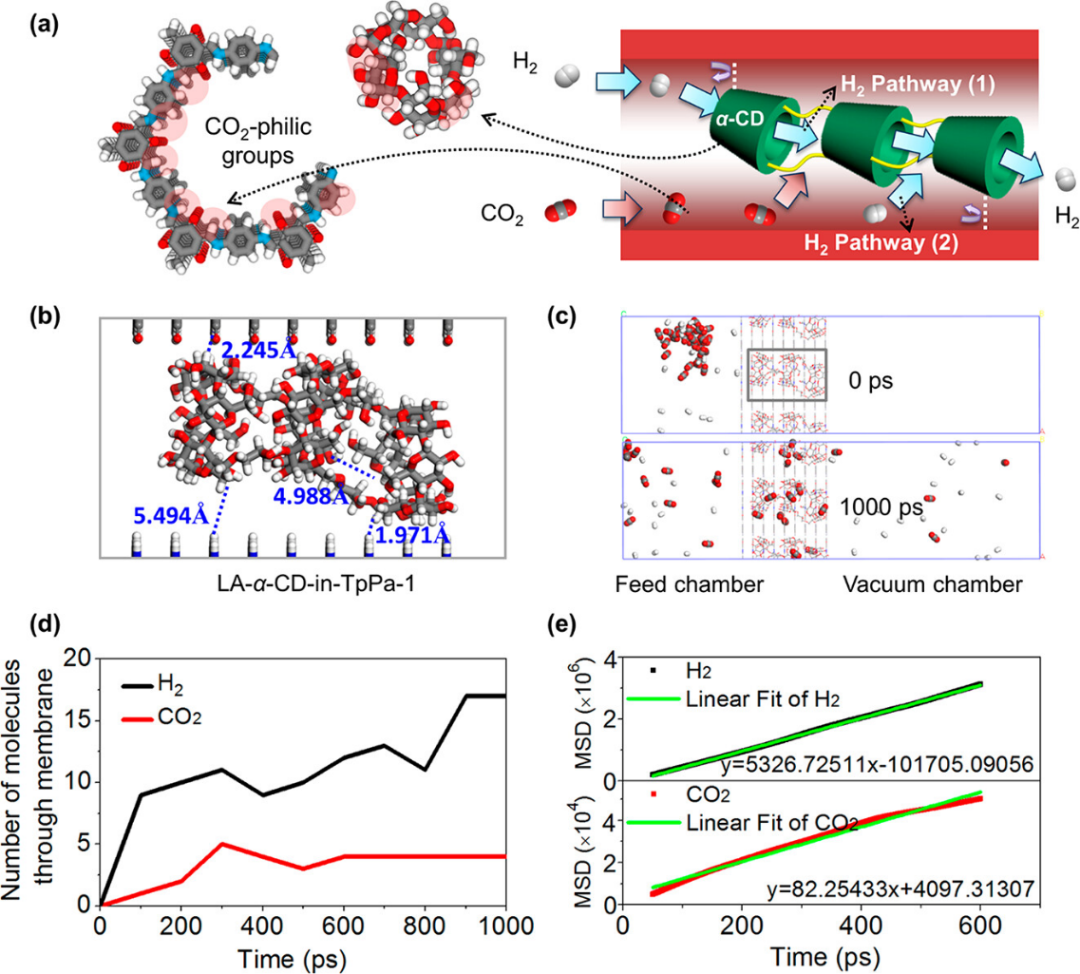
通过分子动力学模拟(MD)深入研究了LA-α-CD-in-TpPa-1膜的气体分离机制,重点分析了H₂和CO₂在膜中的传输行为。
模拟结果清晰揭示了两种不同的气体传输路径(图a和b):对于H₂分子,主要存在两种高效传输通道,第一种是直接通过线性α-CD聚合物内部形成的纳米级孔道(路径1),第二种则是沿着COF主体孔道与α-CD聚合物之间的界面间隙(路径2)。
这两种路径共同构成了H₂分子的快速传输网络。相比之下,CO₂分子由于与膜材料存在更强的相互作用,主要被吸附在COF孔壁的—NH—和—C=O官能团以及α-CD表面的—OH和—C=O位点上,这种强烈的吸附作用不仅显著降低了CO₂的渗透速率,同时也在空间上限制了H₂分子的传输通道。
进一步的定量分析(图c和d)显示,在1000ps的模拟时间内,进料侧的30个H₂分子中有18个(60%)成功渗透至膜的另一侧,而同等条件下的30个CO₂分子仅有4个(13.3%)能够通过膜结构。这一显著的渗透差异直观地证实了该膜材料对H₂的选择性传输特性。
通过对分子运动轨迹的分析和均方位移(MSD)的计算(图e),研究人员发现H₂的扩散系数达到CO₂的65倍,这一结果与实验测得的分离选择性(约35)在趋势上保持一致,虽然绝对值存在一定差异,但这种差异主要源于宏观膜结构与理想模拟模型之间的尺度差异。
这些发现从分子层面阐明了”孔中孔”工程策略的作用机制:通过将线性α-CD聚合物精确嵌入COF纳米孔道中,形成了具有尺寸选择性的超微孔结构。这种结构并非依靠传统的刚性筛分效应,而是通过创造竞争性扩散环境来实现高效的气体分离。具体而言,较大的CO₂分子由于与膜材料的强相互作用而被有效阻滞,而较小的H₂分子则可以利用α-CD内部孔道和界面间隙实现快速传输。
这一机制不仅解释了实验观察到的优异分离性能,也为未来设计更高效的COF基气体分离膜提供了重要的理论指导。研究还指出,通过进一步优化α-CD的含量和排列有序度,有望实现更精确的分子筛分效果,这为后续研究指明了方向。
COF在催化性能研究中的应用
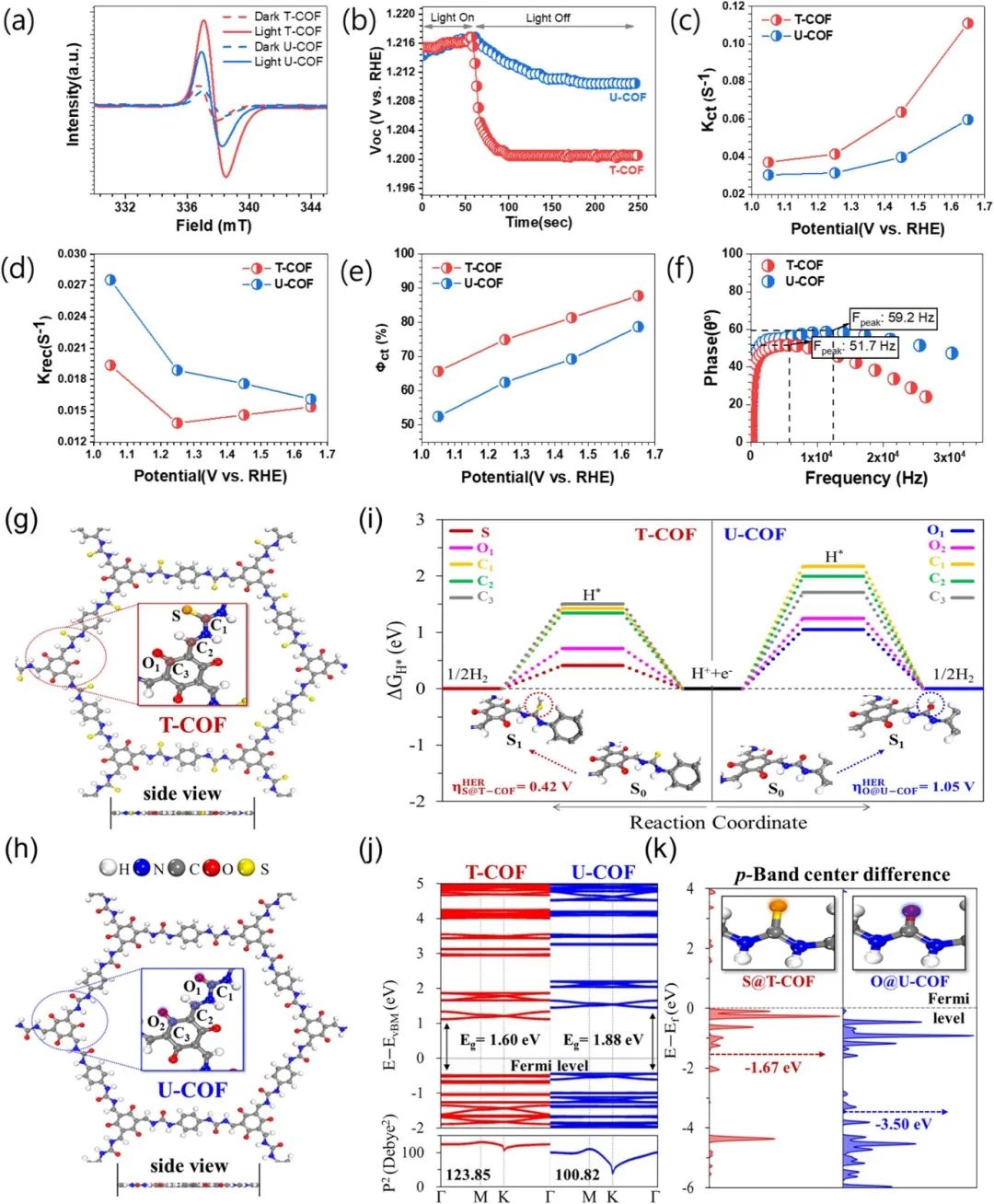
通过构建T-COF(含硫)和U-COF(含氧)的精确理论模型(图g-h),研究不仅验证了实验合成的材料结构合理性,更从原子尺度揭示了硫修饰的关键作用机制。
计算结果表明,T-COF中硫位点表现出接近理想值的氢吸附自由能(ΔG_H*=0.402eV),显著优于U-COF氧位点的1.02eV,这一差异直接解释了实验中T-COF更高的光催化活性(226.4μmol h⁻¹vs.98.2μmolh⁻¹)。
在电子结构层面,DFT计算揭示了硫修饰的多重优化效应:首先,T-COF的带隙缩小至1.60eV(U-COF为1.88eV),使其可见光吸收范围扩展至774nm;其次,导带位置下移至-3.77eV(vs.U-COF的-3.89eV),提供了更强的热力学驱动力;更重要的是,硫的p带中心(-1.67eV)比氧(-3.51eV)更接近费米能级(图5k),显著增强了与氢原子的轨道杂化效率。
综合实验与理论结果,该研究建立了完整的”结构-性能”关系:硫原子通过三重协同机制(能带调控、电荷分离促进、活性位点优化)使T-COF的光电流密度达到4.08 μA cm⁻²,IPCE提升至3.7%,均显著优于U-COF。
这些DFT计算结果不仅解释了实验现象,更为未来设计新型高效COF光催化剂提供了明确的设计原则:通过精准调控杂原子类型(如硫)及其在框架中的位置,可以系统优化材料的能带结构、电荷传输性能和催化活性。