

说明:本文华算科技将从定义出发,系统介绍计算方法、应用实践、关键点,从而帮助读者全面掌握自由能台阶图的解析框架。
自由能台阶图是描述系统(如分子或化学反应)自由能随反应坐标变化的可视化工具,常用于物理化学、生物物理和工程领域,帮助理解能量壁垒和反应路径。
什么是自由能台阶图?


自由能台阶图是一种二维图形,横轴为反应坐标,纵轴为自由能值,通过曲线或曲面展示系统能量最小化路径。其核心是“台阶”概念:每个台阶对应一个亚稳态,台阶高度表示能量壁垒,台阶宽度反映状态稳定性。
自由能台阶图是科学计算中一种核心的可视化工具,用于描绘系统在能量景观中的变化。它通过图形展示自由能(系统可用能量)随反应坐标(如分子构象或反应进度)的起伏,形成类似“台阶”的峰谷结构。
峰代表能量壁垒,谷代表稳定状态,帮助研究者预测反应路径、识别过渡态和优化设计。
DOI:10.1038/s41467-022-35610-w
自由能台阶图的计算
确定反应路径与中间体
在计算自由能台阶图之前,需要明确目标反应的具体反应路径以及其中涉及的关键中间体。不同的化学反应有着不同的反应路径。以二氧化碳还原反应(CO2RR)为例,其可能生成多种产物,如一氧化碳(CO)、甲酸(HCOOH)、甲烷(CH4)等,每一种产物的生成对应着不同的反应路径。
以生成甲烷为例,反应路径可能为CO2→∗COOH→∗CO→∗CHO→∗CH2→∗CH3→CH4,其中∗COOH、∗CO等就是反应过程中的中间体。
确定这些中间体是后续计算的基础,研究者往往需要结合理论知识和实验经验来判断可能的反应路径和中间体。
搭建吸附模型
对于涉及催化剂表面吸附过程的反应,如电催化反应,需要搭建准确的吸附模型。这包括确定催化剂的表面结构、吸附位点以及吸附物种的初始构型。
比如在研究金属催化剂表面的析氢反应(HER)时,要考虑氢原子在金属表面的吸附位点,是顶位吸附、桥位吸附还是穴位吸附。同时,还需考虑催化剂表面与吸附物种之间的相互作用。
密度泛函理论(DFT)计算
在获得优化的中间体结构后,接下来需要计算每个中间体的吉布斯自由能。自由能计算公式为:ΔG = ΔE + ΔZPE – TΔS + eU。
其中:ΔE是DFT计算得到的总能差;ΔZPE是零点能校正;TΔS是熵变项(T为温度,ΔS为熵变);eU为电势修正项(e为电子电荷,U为施加电势)。
自由能校正
单纯的DFT计算得到的能量需要进行校正才能得到准确的吉布斯自由能 。主要考虑以下几个校正项:零点振动能(ZPE)校正、熵校正、溶剂化效应校正(若反应在溶液中进行)、外加电势校正(对于电化学反应)等。
绘制自由能台阶图
将通过上述步骤计算得到的各中间体的吉布斯自由能数据,按照反应路径的顺序进行整理。
使用绘图软件,如Origin、Chemdraw等,以反应坐标为横坐标,吉布斯自由能为纵坐标,将各中间体的自由能数值依次标注在图上,并通过线段将相邻中间体的点连接起来,形成台阶状的曲线。
在图上清晰地标出每个中间体的名称和对应的自由能数值。如果有决速步骤,可以通过特殊的标记,如用阴影或不同颜色突出显示该步骤对应的台阶,使自由能台阶图能够直观地展示反应的能量变化情况。
自由能台阶图的应用
自由能台阶图在催化研究中具有广泛的应用,是连接理论计算与实验研究的桥梁。通过分析自由能台阶图的形态特征,研究人员可以获取催化反应的丰富信息,从而指导催化剂的设计和优化。
催化剂活性评估与设计
自由能台阶图最直接的应用是评估催化剂的活性和识别决速步骤。决速步骤是指反应路径中能垒最高的步骤,它决定了整个反应的反应速率。在自由能台阶图中,ΔG的峰值对应决速步。
例如,在CO2还原反应中,若*COOH → *CO步骤的ΔG为全路径最高,则该步骤为RDS。这一信息为催化剂优化提供了明确方向。
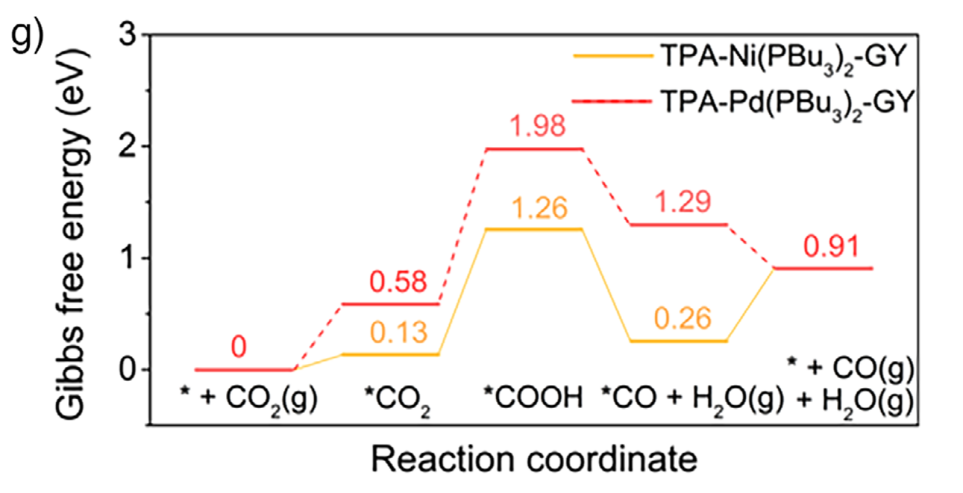
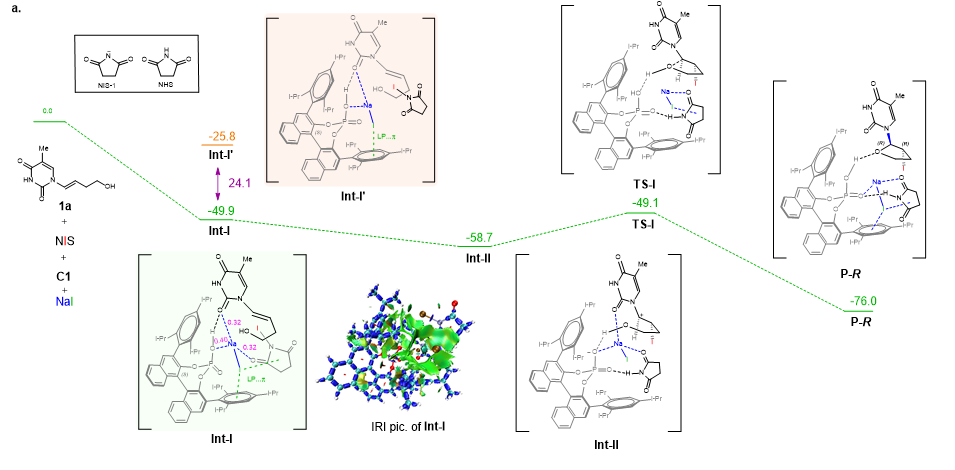
图1 CO2还原反应台阶图
DOI:10.1038/s41467-022-35610-w
催化剂设计的核心在于优化中间体在催化剂表面的吸附强度。理想的催化剂应该与反应中间体具有适中的吸附强度——太强会导致中间体难以进一步反应,太弱则不利于反应的初始活化。自由能台阶图可以直观展示不同催化剂表面对中间体吸附强度的影响。
反应选择性预测
除了评估催化活性外,自由能台阶图还可以预测反应的选择性。
在多电子转移反应中,如CO2还原反应,往往存在多条竞争路径,生成不同产物(如CO、HCOOH、CH4等)。通过绘制不同路径的自由能台阶图,并对比它们的决速步能垒,可以预测反应的主要产物。
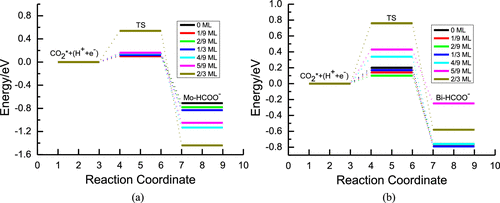
图2 CO2还原反应自由能台阶图
DOI:10.1021/acsomega.0c00227
例如,若生成CO的RDS(*COOH →*CO)ΔG为+0.7 eV,而生成HCOOH的RDS(*OCHO→HCOOH)ΔG为+1.0 eV,则反应更倾向于生成CO,因为其能垒较低。这种选择性预测对于工业应用至关重要,因为通常需要高选择性地生成特定有价值产物。
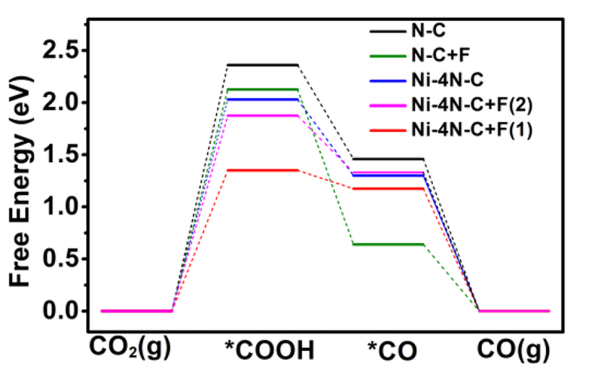
图3 CO还原反应自由能台阶图
DOI:10.1016/j.apcatb.2020.119591
自由能台阶图还可以分析电位依赖性的选择性变化。通过调节施加电势(U),可以改变各步骤的ΔG(ΔG = ΔG₀ + eU),进而调控产物选择性。
例如,在低电位下,CO2可能更易还原至COOH;高电位下则推动CO进一步还原。这为实验中选择最佳电位窗口提供了理论指导。
实验验证与机理研究
自由能台阶图不仅是理论工具,更是连接计算与实验的桥梁。通过对比理论预测与实验结果,可以深入解析催化反应的微观机制。
当实验发现某催化剂活性异常时,可通过自由能台阶图分析其决速步ΔG的变化,从而解释异常活性的来源。
刊案例详解:氟调谐单原子镍催化剂实现高效CO2电还原
研背景与核心创新
本研究通过氟掺杂策略调控镍单原子催化剂的电子结构,成功实现了高效CO2-to-CO转化,法拉第效率>95%,CO生成速率达1146 mmol g-1 h-1。
自由能台阶图揭示机理
为了理解氟掺杂对催化性能的影响机制,研究人员绘制了自由能台阶图对比分析掺杂前后的变化。计算结果显示,在未掺杂的镍催化剂上,*COOH形成步骤的能垒较高(ΔG = +0.85 eV),表明这一步骤是决速步。
而在氟掺杂催化剂上,这一步骤的能垒显著降低(ΔG = +0.62 eV),从而提高了整体反应速率。
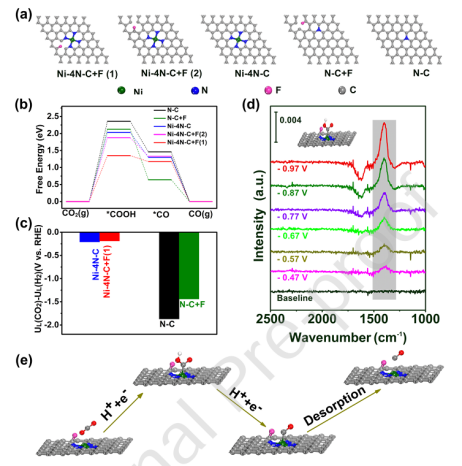
图4 CO2-to-CO的DFT理论研究
DOI:10.1016/j.apcatb.2020.119591
自由能台阶图进一步揭示了氟掺杂的电子效应:X射线光电子能谱(XPS)显示,F掺杂后Ni的2p轨道结合能正移0.3 eV,表明电子从Ni原子向F原子转移,优化了Ni活性中心对*COOH中间体的吸附强度。
这种电子重分布效应使反应路径上的各中间体吸附能更加均衡,避免了某个步骤能垒过高,从而提升了整体催化性能。
理论计算与实验验证
研究展示了如何通过理论计算与实验验证相结合深入探究催化机理。除了自由能台阶图分析,研究人员还进行了d带中心计算,发现F掺杂后Ni的d带中心下移,减弱了对反应中间体的过强吸附,使反应更容易进行。
这些理论预测通过原位光谱实验得到了验证。利用原位X射线吸收谱(XAS)和红外光谱,研究人员直接观测到了反应中间体的形成和转化过程,证实了F掺杂确实优化了*COOH中间体的吸附行为,与自由能台阶图预测的结果一致。
这项研究体现了自由能台阶图在理性催化剂设计中的关键作用:通过分析不同改性策略对自由能台阶图的影响,可以预测催化性能变化趋势,指导实验方向,避免盲目尝试。这种研究模式已经成为顶级期刊发表催化研究工作的标准范式。
总结
自由能台阶图作为催化研究中不可或缺的工具,通过图形化展示反应路径中的能量变化,为理解反应机理、评估催化剂性能和指导催化剂设计提供了强大支持。
从定义上看,自由能台阶图是描述反应路径中各步骤吉布斯自由能变化的可视化工具;从计算方法上看,它基于DFT计算,包括中间体结构优化、自由能计算等步骤;从应用上看,它在催化剂活性评估、选择性预测和实验验证等方面发挥着重要作用。
通过本文的论述和案例剖析,我们可以看到自由能台阶图已经成为连接理论计算与实验研究的桥梁,在顶级研究中占据核心地位。